“Lässt du dich gegen SARS-CoV-2 impfen?” Diese Frage wurde mir in den letzten Wochen mehrfach gestellt und meine Antwort lautet: ja. Sobald alle gefährdeten Gruppen geimpft sind, werde ich mich impfen lassen. Ich möchte hier kein Plädoyer für das Impfen schreiben oder mich mit den Fürs und Widers auseinandersetzen. Das wurde sicher oft genug an anderer Stelle getan. Ich bin auch keine Vakzinologin, Immunologin oder Mikrobiologin und kann großartig mit Impfwissen auftrumpfen.
Stattdessen möchte ich euch lieber auf eine bioinformatische Reise mitnehmen, und den genetischen Inhalt des Impfstoffes genau unter die Lupe nehmen. Ich bin über diese wirklich großartige Zusammenfassung von Bert Hubert (übersetzt von Friedrich Zahn) gestolpert, der die fantastischen Tricks und Kniffe beschreibt, die für die Wirksamkeit des Impfstoffes sorgen. Und beim Lesen des Beitrags wurde mir einmal mehr bewusst: “Wissenschaft ist sooooo cool!”
Vermutlich habt ihr bereits mitbekommen, dass es sich bei dem BioNTech/Pfizer SARS-CoV-2 Impfstoff um einen mRNA Impfstoff handelt. mRNA steht kurz für messenger RNA (im Deutschen auch Boten-RNA genannt). Eine mRNA ist sozusagen eine Kopie, die von einem Gen auf unserer DNA gemacht wird, um die Information aus dem Zellkern (wo sich die DNA befindet) in die Zelle transportieren zu können, wo sie als Bauanleitung für ein bestimmtes Protein fungiert.
Bert Hubert nutzt in seinem Beitrag einen schönen Vergleich: Er beschreibt die DNA als Festplattenspeicher unseres Körpers: widerstandsfähig, redundant, ausfallsicher, zuverlässig. Die RNA hingegen ist die flüchtige “Arbeitsspeicher”-Variante der DNA: schneller und vielseitiger, aber eben auch fragiler und anfälliger. Daher muss der Pfizer/BioNTech mRNA Impfstoff bei Tiefsttemperaturen gelagert werden. Andererseits haben mRNA Impfstoffe den Vorteil, dass eine große Anzahl Impfdosen innerhalb weniger Wochen hergestellt werden kann.
Das nötige Hintergrundwissen
Bioinformatiker sehen DNA und RNA ja nicht als komplexe Moleküle, sondern als simple Zeichenketten. Und deswegen werden wir die Zeichenkette des mRNA Impfstoffs (und was sie so besonders macht) genauer unter die Lupe nehmen. Für ein wenig Hintergrundwissen über DNA, RNA, Nukelobasen, Proteine und Aminosäuren empfehle ich euch erst noch diesen Beitrag zu lesen. Kurz zusammengefasst solltet ihr folgendes wissen:
- DNA/RNA sind Kettenmoleküle aus Nukleotiden.
- Diese Nukleotide stellen wir als die Zeichen A,C,G und T (in der DNA) bzw U (in der RNA) dar.
- Proteine sind Kettenmoleküle aus Aminosäuren.
- Es gibt 20 verschiedene Aminosäuren.
- Jeweils 3 Nukleotide (zusammen als Codon bezeichnet) werden in eine Aminosäure übersetzt.
- Das sind 43 = 64 mögliche Codons (und damit mehr als 20 verschiedene Aminosäuren).
- Das heißt, es gibt für fast alle der Aminosäuren mehrere verschiedene Codierungen.
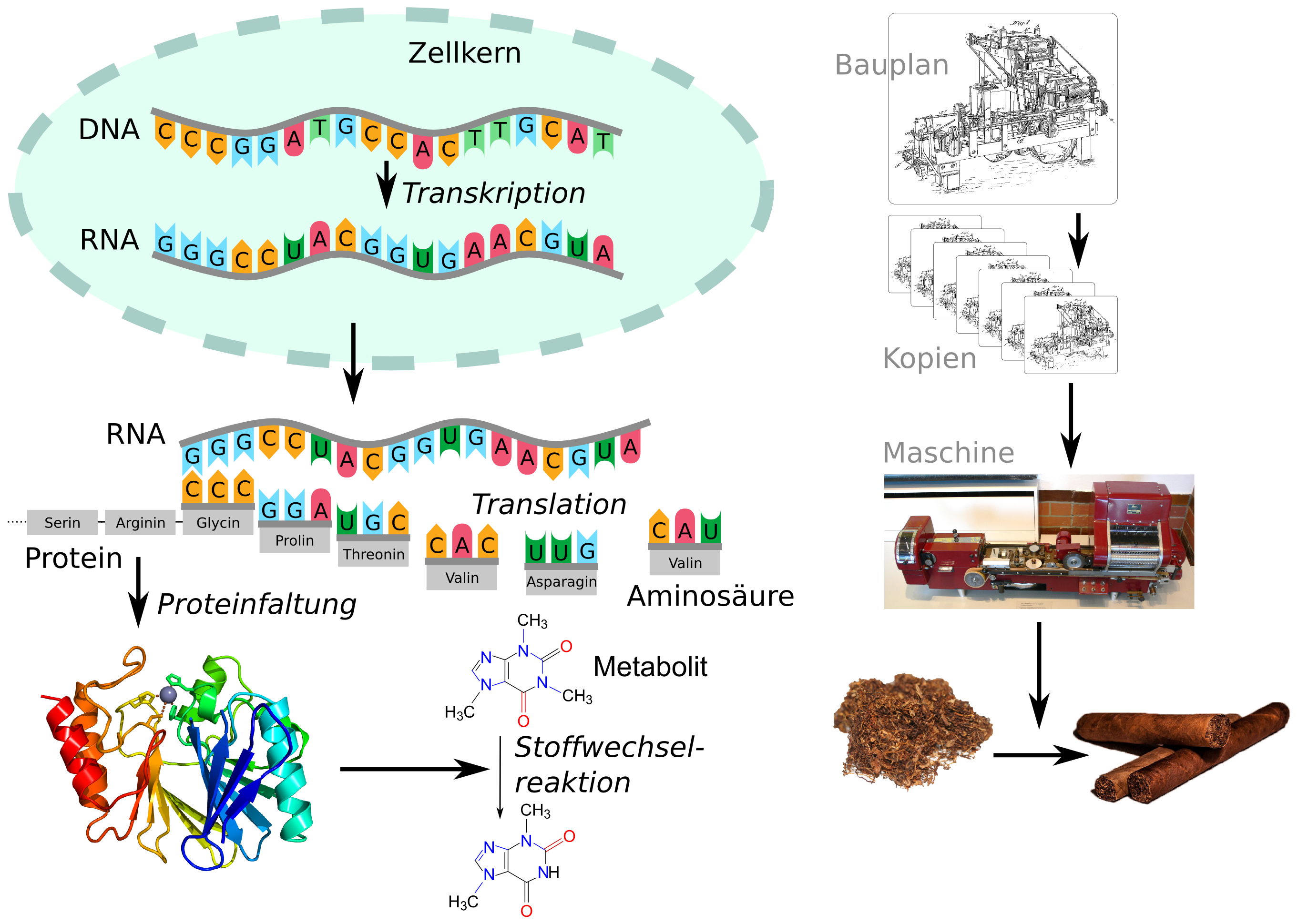
Vom Gen zum Protein
Die Idee hinter mRNA als Impfstoff
Ziel einer aktiven Schutzimpfung ist es, dem Immunsystem des Körpers beizubringen, auf einen Krankheitserreger (Virus oder Bakterium) schnell und wirksam zu reagieren. Das geschieht in der Regel, indem wir unseren Körper mit einem geschwächten oder entschärften Erreger konfrontieren, an dem das Immunsystem die Abwehrreaktion trainieren kann. Werden wir dann mit dem tatsächlichen Erreger infiziert, erkranken wir im besten Fall gar nicht oder nur sehr leicht.
Auch eine mRNA-Impfung trainiert das Immunsystem gegen den Erreger, aber auf eine besonders gezielte und leistungsstarke Weise. Wir müssen keinen geschwächten oder entschärften Erreger spritzen, stattdessen verabreicht man mRNA, die den genetischen Code für ein bestimmtes Protein enthält. Bei diesem Protein handelt es sich um ein sogenanntes Antigen, das eine Immunantwort des Körpers auslöst. Und genau diese Immunantwort soll unser Körper trainieren. Unser Körper lernt, genau die Erreger zu bekämpfen, die solche Antigene auf ihrer Oberfläche tragen. Die RNA selbst erzeugt keine Immunreaktion und wird nach kurzer Zeit in der Zelle abgebaut.
Ein kurzer Exkurs in die Immunabwehr
Für eine gezielte Immunreaktion gegen einen bestimmten Erreger sind unter anderem B-Lymphozyten zuständig. Es gibt unzählige verschiedene B-Lymphozyten mit unterschiedlichen Bindungsstellen an ihrer Oberfläche. Zu den Antigenen auf der Oberfläche eines bestimmten Erregers passen nur einige wenige B-Zellen. Durch Bindung an dieses spezifische Antigen werden die B-Lymphozyten aktiviert, sich zu vermehren. Die Zahl der B-Lymphozyten, die den Erreger erkennen können, nimmt also stark zu. Ein Teil der B-Lymphozyten reift zu Plasmazellen, die Antikörper speziell gegen den Erreger produzieren. Diese Antikörper sind Proteine, die spezifisch an das bestimmte Antigen binden. Sie machen Krankheitserreger unschädlich, indem sie beispielsweise Bakterien miteinander verkleben oder Viren die Fähigkeit nehmen, in Körperzellen einzudringen und sich dort zu vermehren. Außerdem werden Erreger, die mit Antikörpern markiert sind, leichter von der Immunabwehr aufgespürt.
Bis unser Immunsystem diese Immunantwort gegen einen bestimmten Erreger gelernt hat, kommt es zu Krankheitsbeschwerden. Beim ersten Kontakt, bilden sich erst nach einigen Tagen Antikörper. Nach erfolgreicher Bekämpfung des Erregers nimmt deren Konzentration wieder ab. Ein Teil der B-Lymphozyten entwickelt sich jedoch zu Gedächtniszellen, die für Jahre oder manchmal lebenslang erhalten bleiben. Bei einem erneuten Kontakt mit dem gleichen Erreger setzt die Produktion der Antikörper viel schneller ein und erreicht größere Mengen. Die Immunreaktion ist gezielter und schneller; Krankheitserscheinungen sind schwächer oder treten gar nicht erst auf.
Fun fact:
Das Wort Vakzination bzw Vakzinierung stammt vom lateinischen Wort für Kuh (vacca) ab. Ab Ende des 18. Jahrhunderts wurde der Wirksamkeitsnachweis einer Pockenimpfung mit dem Vacciniavirus erbracht (statt mit menschlichen Pockenviren als Lebendimpfstoff), die seltener zu einer Erkrankung führte. Das Vacciniavirus wurde ursprünglich für ein Kuhpockenvirus gehalten. Inzwischen ist bekannt, dass das Vacciniavirus näher mit den Pferdepocken als mit den Kuhpocken verwandt ist.
Der SARS-CoV-2 Impfstoff

Struktur eines Coronavirus: SARS-CoV-2 ist gespickt mit Spike-Proteinen (rosa) die vom menschlichen Immunsystem als Antigene erkannt werden. SARS-CoV-2 nutzt diese Proteine, um an menschliche Zellen anzudocken und sein Erbgut ins Zellinnere zu entlassen. (Creative Commons Attribution-Share Alike 4.0 International; by Manu5)
Im Falle von SARS-CoV-2 trainieren wir gezielt gegen das berühmt-berüchtigte Spike-Protein. Die Verpackung des SARS-CoV-2 ist mit Spike-Proteinen gespickt. Es nutzt diese Proteine, um an menschliche Zellen anzudocken. Das Virus kann dann mit der Zellmembran verschmelzen und sein Erbgut ins Zellinnere entlassen. Das Spike-Protein ist das Antigen, dass unser Körper kennenlernen soll. Der Impfstoff enthält mRNA, der dieses Spike-Protein beschreibt. Und zwar nur dieses Protein und nicht das gesamte Virus. Durch clevere chemische Tricks gelangt der Impfstoff in einige unserer Zellen. Diese produzieren das Spike-Protein in großen Mengen. Unser Immunsystem kann an diesem Protein trainieren und Gedächtniszellen entwickeln. Werden wir dann mit SARS-CoV-2 infiziert, sind wir für eine schnelle und gezielte Abwehrreaktion gewappnet.
Ein Blick in den genetischen Code des Impfstoffes
Jetzt schauen wir uns die 4284-Zeichen-lange Kette des mRNA Impfstoffs im Detail an. Die komplette Zeichenkette könnt ihr bei der WHO einsehen. Die mRNA besteht aus mehreren Abschnitten, die wir uns im Folgenden näher angucken.
Kappe
Der Code des Impfstoffes startet mit den folgenden zwei Nukleotiden (als Kappe bezeichnet): GA. Diese Kappe sorgt dafür, dass die mRNA von den Ribosomen erkannt wird. Ribosomen sind quasi die “Übersetzer”, die die Nukleotid-Zeichenkette in eine Aminosäure-Zeichenkette übersetzen und dabei das Protein “zusammenbauen” (quasi eine Art 3D-Drucker für Proteine). Die Kappe erhöht außerdem die Halbwertszeit der mRNA. Je langsamer die mRNA in der Zelle abgebaut wird, desto häufiger kann sie übersetzt und desto mehr Protein hergestellt werden.
Leitsequenz
Es folgt die sogenannte Leitsequenz oder 5′-UTR. UTR steht für untranslated region, also ein nicht-übersetzter Abschnitt. Dieser Teil wird nicht in Aminosäuren übersetzt und landet damit nicht im Protein. Dieser Abschnitt dient zur Steuerung der Herstellung des Proteins. Er enthält zum Beispiel die Bindungsstelle für das Ribosom. Das Ribosom muss physisch in Kontakt mit dem RNA-Strang sein, damit die Herstellung des Proteins funktionieren kann. Diese Bindungsstelle heißt Kozak-Sequenz, benannt nach der US-amerikanischen Biochemikerin Marilyn Kozak.
Zusätzlich enthält die Leitsequenz übergeordnete Informationen, etwa wann und wie oft die Übersetzung in Proteine geschehen soll. Für den Impfstoff wurde die “dringlichste” bekannte Version der Leitsequenz gewählt, basierend auf einem Gen, das dafür bekannt ist, dass es zuverlässig eine große Zahl an Proteinen herstellt.
Die Leitsequenz des mRNA Impfstoffes sieht folgendermaßen aus:
GAAΨAAACΨAGΨAΨΨCΨΨCΨGGΨCCCCACAGACΨCAGAGAGAACCCGCCACCEs fällt auf: Statt den für RNA üblichen vier Nukleinbasen A, C, G und U finden wir statt des U’s ein Ψ. Dies ist eine der außergewöhnlich schlauen Lösungen des Impfstoffs. Unsere Zellen sind extrem skeptisch gegenüber fremder oder synthetischer RNA and setzen alles daran, diese zu zerstören, bevor sie ihre Zielzellen erreichen. Damit sich die mRNA am Immunsystem vorbeimogeln kann, verwendet man ungewöhnliche Nukleoside. Der Impfstoff enthält eine minimal veränderte Form des Us, nämlich 1-Methyl-3’-Pseudouridylyl, dargestellt als Ψ. Damit kann die mRNA nicht von RNAsen abgebaut werden und ein Immunangriff wird unterdrückt. Alle für die Herstellung des Proteins relevanten Teile der Zelle erkennen das Ψ aber als reguläres U an.
Hier möchte ich gleich einem der vielen Aufschreie gegen diesen Impfstoff entgegenargumentieren: Oft höre ich die Frage “Wie kann es sein, dass der Impfstoff in so kurzer Zeit entwickelt wurde?”. Nun, wir ernten hier die Früchte der in der Vergangenheit geleisteten wissenschaftlichen Grundlagenforschung. Viele der verwendeten “Tricks” sind nicht neu und wurden schon vorher entdeckt und erforscht. An dieser modifizierten Art der mRNA zum Beispiel wird schon seit Jahren insbesondere für die Regeneration von geschädigtem Herzmuskelgewebe geforscht. Die Entdecker:innen der Ψ-Ersetzungsmethode Katalin Karikó und Drew Weissman mussten etwa 15 Jahre dafür kämpfen, dass ihre Forschung finanziert und akzeptiert wurde, weil man den weitreichenden Nutzen damals noch nicht absehen konnte.
Signalpeptid
Der nächste Abschnitt der mRNA enthält die Information zur Herstellung des Signalpeptids. Als Peptide bezeichnet man kurze Aminosäure-Ketten (zum Beispiel Teilstücke von Proteinen). Das Signalpeptid ist das erste Teilstück des Spike-Proteins und entscheidet über dessen Bestimmungsort. Es ermöglicht, dass das Protein (über das Endoplasmatische Retikulum) aus der Zelle hinaus transportiert werden kann, um dort von unserem Immunsystem erkannt zu werden und den Lernprozess unseres Immunsystems zu starten.
Das Signalpeptid ist nicht sehr lang. Schauen wir uns die Zeichenketten der Impstoff-RNA im Vergleich zur echten Virus-RNA an. Die Zeichenkette ist in Dreiergruppen gegliedert: Drei Nukleotide sind ein Codon und werden in eine Aminosäure übersetzt.
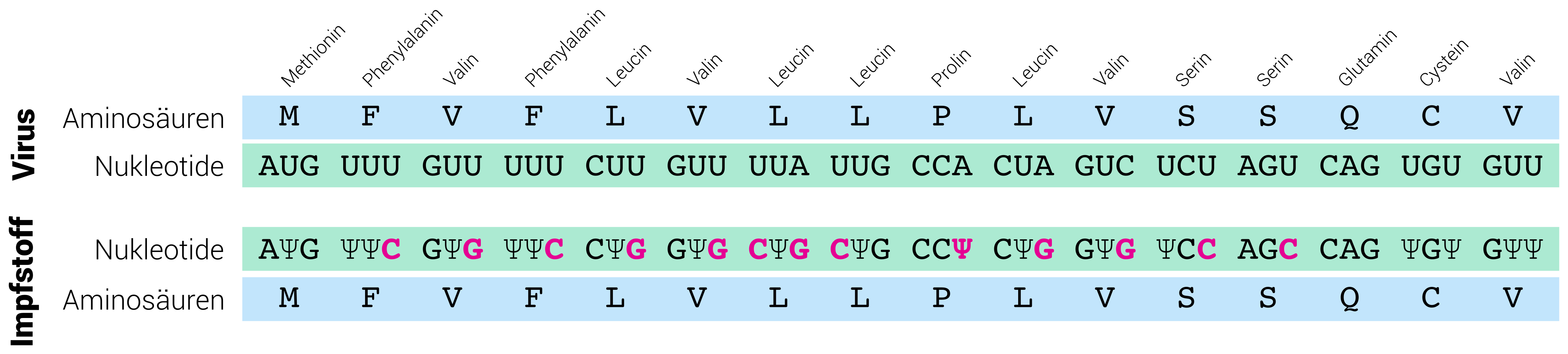
Wir sehen, dass sich die RNAs an manchen Stellen unterscheiden. Die resultierenden Aminosäuren bleiben jedoch unverändert. Das Signalpeptid im Impfstoff besteht aus genau den gleichen Aminosäuren wie im Virus selbst. Wie kann sich dann die RNA unterscheiden? Und wozu hat man diese Unterschiede in die Impfstoff-RNA eingefügt?

Genetischer Code: Jeweils drei Bausteine der RNA lassen sich in eine Aminosäure übersetzen.
Wie oben erwähnt, gibt es 64 mögliche Dreierkombinationen von Nukleotiden und damit mehr als die 20 verschiedenen Aminosäuren. Das heißt, es gibt für fast alle der Aminosäuren mehrere verschiedene RNA-Codons (Synonyme könnte man sagen). Oft unterscheiden sich die Codons für eine Aminosäure nur im letzten Nukleotid. Zum Beispiel wird sowohl CCU, CCG, CCA und CCC in die Aminosäure Prolin übersetzt. Auch alle im Impfstoff vorgenommenen Ersetzungen sind Synonyme. Die Aminosäuren verändern sich nicht. Wozu dann überhaupt Änderungen einfügen? Auch hier geht es wieder um Effizienz: RNA, die viele C’s und G’s enthält, wird effizienter in Proteine verwandelt. Zu diesem Zweck wurden in der Impfstoff-RNA möglichst viele Zeichen durch C’s und G’s ersetzt.
Das Spike-Protein
Die nächsten 3777 Zeichen der Impfstoff-RNA enthalten den genetischen Code (also den Bauplan) für die Herstellung des Spike-Proteins. Sie sind in ähnlicher Weise optimiert, das heißt, es wurden möglichst viele C’s und G’s eingefügt ohne die resultierende Aminosäure-Kette zu verändern. Das gilt für alle, bis auf zwei Aminosäuren des Proteins.
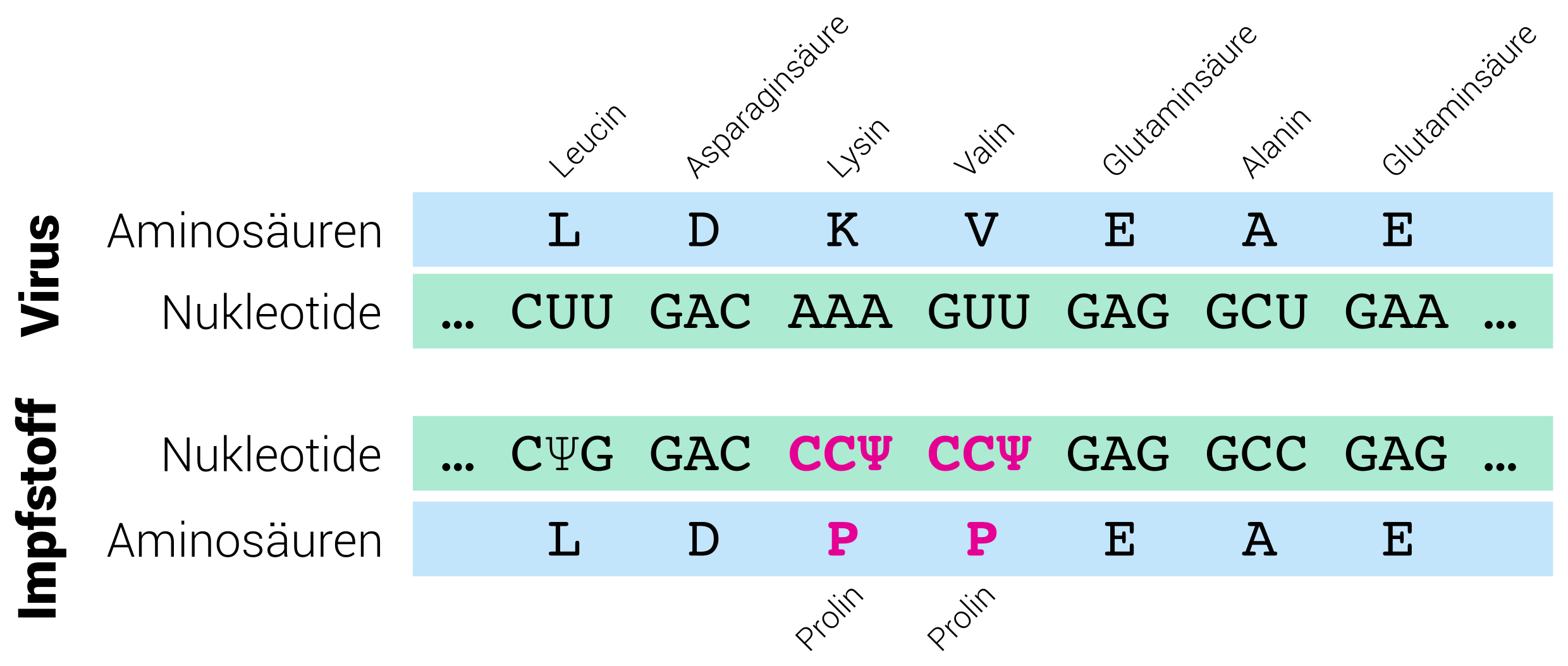
Zwei der Aminosäuren wurden durch eine andere Aminosäure ersetzt (nämlich Prolin). Der Grund liegt verborgen in der 3D-Struktur (oder Raumstruktur) des Proteins. Bis hier haben wir Proteine als Aminosäure-Kettenmoleküle betrachtet. Tatsächlich faltet sich diese Kette (2D-Struktur) aber zu einer 3D-Struktur. Vergleichbar mit einem Faden, den ihr zusammenknüllt. Die räumliche Form des Spike-Proteins ähnelt einem Stachel oder Dorn (engl. spike). Diese Form nimmt das Protein jedoch nur an, wenn es in die Virus-Verpackung (als Kapsid bezeichnet) verbaut wird. Die Idee des Impfstoffs ist es aber (wie oben erwähnt), nicht das ganze Virus oder Kapsid zu erzeugen, sondern nur das eine spezifische Protein, das als Erkennungsmerkmal des Virus ausreicht. Das Problem an der Sache: ein freies Spike-Protein nimmt eine völlig andere Form an. Unser Körper würde also ein falsches Erkennungsmerkmal lernen und den eigentlichen “Täter” später nicht erkennen.
Auch hier haben wir riesiges Glück, dass wir auf bereits vorangegangen Forschungsarbeiten zurückgreifen konnten. 2017 veröffentlichten Forscher:innen, dass das Ersetzen zweier bestimmter Aminosäuren dazu führt, dass das Spike-Protein (damals schon aus SARS-CoV-1 und MERS bekannt) seine Stachelform einnimmt, auch ohne Teil der Virus-Verpackung zu sein. Somit kann unser Immunsystem allein auf diesem Protein lernen, woran es den Eindringling erkennt, und im Ernstfall schnell und effizient reagieren.
Der Rest
Die Impfstoff-mRNA endet mit der sogenannten 3′-UTR gefolgt vom Poly(A)-Schwanz. Die 3′-UTR hat oft regulatorische Funktionen und sorgt auch wieder für RNA Stabilität und eine möglichst große Menge an produziertem Spike-Protein. Der Poly(A)-Schwanz ist (wie der Name sagt) eine lange Kette von A’s, mit der die mRNA endet. mRNA kann immer wieder verwendet werden, verliert dabei jedoch stets einige A’s am Ende. Sind diese aufgebraucht, wird die mRNA abgebaut und entsorgt. Der Poly(A)-Schwanz schützt also einerseits vor vorzeitigem Verschleiß, sorgt aber andererseits auch dafür, dass die mRNA schlussendlich durch so genannte Ribonucleasen (Enzyme, die RNA abbauen) entsorgt wird.
Von der Buchstabenkette zum Impfstoff
Wir haben uns jetzt aus informatischer Sicht die Zeichenkette der mRNA angeschaut und analysiert, was diese Zeichen bedeuten. Wie wird aus dieser Zeichenkette eine Flüssigkeit, die ich mir spritzen lassen kann? Zu Beginn der Impfstoffproduktion wurde dieser Code an einen DNA-Drucker geschickt, welcher aus der Zeichenkette echte DNA-Moleküle herstellt. Aus der Maschine kommt eine winzige Menge DNA, die (durch biochemische Methoden) in RNA umgewandelt wird. Um die Aufnahme durch Körperzellen zu ermöglichen, wird die mRNA mit einer cleveren Lipid-Verpackung umhüllt. Fertig ist der Impfstoff.
Warum der mRNA Impfstoff nicht eure DNA verändern wird
Eine gängige Sorge, die oft geäußert wird, ist die Frage, ob die mRNA unser Genom verändern kann. Tatsächlich würde das gegen alles verstoßen, was wir über Zellbiologie wissen. Die Kurzversion: mRNA-Impfstoffe wirken vollständig im Zytosol der Zelle und kommen nicht in die Nähe des Zellkerns, wo sich die DNA befindet. Die mRNA hat also keinen Einfluss auf die menschliche DNA, weder in Körperzellen noch in Keimbahnzellen. Sie wird (wie oben beschrieben) nach kurzer Zeit abgebaut. Nach dem Abbau der mRNA kann auch das Spike-Protein nicht weiter hergestellt werden. Was bleibt sind die Gedächtniszellen unseres Immunsystems, die nun dazu in der Lage sind, den Eindringling schnell zu erkennen und effizient zu bekämpfen. Die ausführliche Erklärung eines Mikrobiologen könnt ihr hier nachlesen. Wenn ihr trotzdem wegen des mRNA-Impfstoffes besorgt seid, hier die gute Info: Es ist sehr wahrscheinlich, dass es bald auch andere Arten von Impfstoffen gegen SARS-CoV-2 geben wird.
Für weitere Fragen zur SARS-CoV-2 Impfung haben das RKI und das Bundesministerium für Gesundheit Antworten zu den gängigen Fragen bereitgestellt.
![]()






















































{kind=link}