
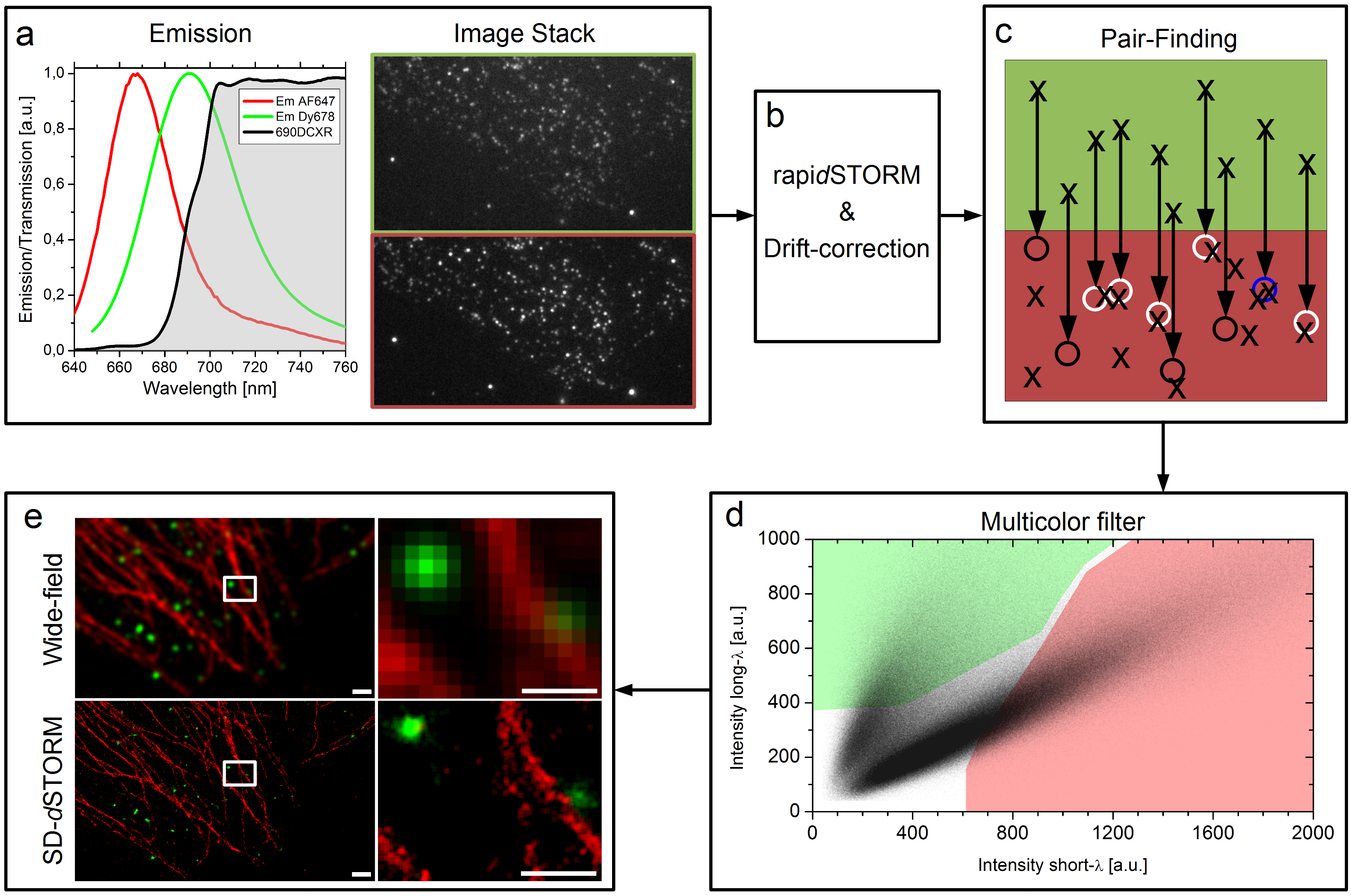
Unsere Idee war es dieses Problem komplett zu umgehen in dem wir zwei Farbstoff genommen haben die recht nahe beieinander liegen, sich aber in ihrem Spektrum ein bisschen unterscheiden. Wir haben zwei rote Farbstoffe verwendet, die man mit dem Auge so gut wie gar nicht unterscheiden kann. Dann haben wir einen Spiegel vor die Kamera gesetzt, der bis zu einer bestimmten Wellenlänge das Licht passieren lässt aber oberhalb dieser Wellenlänge anfängt das Licht zu reflektieren. So entstehen zwei Bilder unserer Probe, die wir nebeneinander auf einer Kamera abgebildet haben. So sollten also von jedem Farbstoff ein Licht-Punkt-Paar entstehen, auf jeder Seite der Kamera ein Licht-Punkt pro Blink-Ereignis. Man kann das im unteren Bild im Kasten a) sehen. Von diesen Bildern haben wir normalerweise zehn bis zwanzigtausend Bilder pro Probe aufgenommen und dann mit einer Software die Positionen der einzelnen Licht-Punkte errechnen lassen. Die Software die wir dafür verwendet haben heißt rapidSTORM und ist kostenlos und open source.
Der Workflow von SD-dSTORM. Lampe et al., Spectral demixing avoids registration errors and reduces noise in multicolor localization-based super-resolution microscopy, Methods and Applications in Fluorescence, Volume 3, Number 3, 13 August 2015 © IOP Publishing. Reproduced with permission. All rights reserved
Jetzt haben wir also einen Haufen von Punkten, oder besser gesagt haben wir Punkte-Paare, denn aus einem Blink-Ereigniss wurde durch den Spiegel vor der Kamera ein Licht-Punkte-Paar. Dieses Paar sagt uns direkt etwas über die Farbe, denn der hellrote Farbstoff (kürzere Wellenlänge) sollte auf der einen Seite einen helleren Lichtpunkt hinterlassen haben als auf der anderen, wohingegen der dunkelrote Farbstoff (längere Wellenlänge) sich genau anders herum verhalten sollte. Also müssen wir jetzt nachträglich aus der langen Liste von Positionen unsere Paare ausfindig machen. Das sieht man im Bild oben in Kasten c). Wie die Geometrie unserer Kamera und des Spiegels davor ist wissen wir, also wissen wir auch in welchem Abstand wir suchen müssen. Da aber keine Optik perfekt ist, suchen wir nach einem Partner in einer kleinen Umgebenung, was durch kleine Kreise am Ende der Pfeile angedeutet ist. Wenn da ein Blink-Ereignis im gleichen Bild statt gefunden hat, dann haben wir ein Paar gefunden, wenn wir kein Ereignis finden wird die ursprüngliche Position verworfen und wenn wir mehr als einen Partner finden können wir uns nicht mehr sicher über die Farbe sein und verwerfen alle beteiligten Lokalisationen. Das mag einem seltsam vorkommen, dass wir hier Daten “weg werfen”, aber das ganze hat auch einen Vorteil, wie man später noch sehen wird.
Am Ende haben wir also eine Liste von Paaren, immer jeweils zwei Lokalisationen, also auch zwei mal eine Helligkeitsinformation, einmal aus dem Kanal bei dem der Spiegel eine kürzere Wellenlänge durchgelassen hat und einem aus dem Kanal bei dem der Spiegel eine längere Wellenlänge reflektiert hat. Diese beiden Helligkeiten eines jeden Paars stellt man in einem Graphen da, wie man oben in Kasten d) sehen kann. Dort erkennt man schon zwei getrennte Populationen von Punktwolken, jeder Punkt steht für ein Paar. Und jetzt werden schon wieder Daten weggeworfen. Nur die Paare, die in einem der farbigen Bereiche liegen werden einem Farbstoff zugeordnet. Etwas irritierend hierbei unsere Farbenwahl: Der hellrote Farbstoff wird hier in rot dargestellt, der dunkelrote Farbstoff in grün. Längst nicht alle Paare liegen innerhalb der farbigen Flächen in d) und diese Paare werden auch weggeworfen. Wir machen das, weil man sich nicht überall in diesem Graphen sicher sein kann, dass dort definitiv nur Paare von dem einen Farbstoff gefunden werden und auf keinen Fall Paare von dem anderen. Das schlimmste was einem passieren kann ist einfach, dass man Lokalisationen eine falsche Farbe zuweist. Mit diesem “Multicolor filter” schaffen wir es die Falschzuweisung von Farben auf unter 1% zu drücken. Wie dann am Ende des rekonstruierte Bild aussieht, auch im Gegensatz zu einem Bild aus einem normalen Fluoreszenzmikroskop, sieht man oben in Kasten e). Warum es gar nicht verkehrt ist auch mal Daten weg zu werfen, sieht man im nächsten Bild.
 orcid.org/0000-0003-0750-4757
orcid.org/0000-0003-0750-4757
Kommentare (8)