
Ich muss grinsen beim Schreiben dieser ersten Zeilen. Hochauflösungsmikroskopie ist mein Ding, ich habe in dem Bereich meine Dokorarbeit geschrieben, ich habe viele Freund*innen und Kolleg*innen, die in dem Bereich forschen – ach ich mag das Feld einfach, es hat mich von Anfang an fasziniert. Aber was das jetzt genau ist verrät dieses Wortungetüm leider noch nicht direkt, und eigentlich ist diese lange Aneinanderreihung von Buchstaben eigentlich auch nur eine verkürzte Bezeichnung. Richtig müsste es eigentlich Hochauflösungsfluoreszenzmikroskopie heißen, nur nennt es so niemand. Meistens spricht man von Super-Resolution als Oberbegriff, nicht nur weil die englische Bezeichnung irgendwie schmissiger klingt, sondern auch weil man in den meisten Fällen sowieso die englischen Begriffe im Kopf hat.
Also: Was ist Super-Resolution?
Der ein oder andere erinnert sich vielleicht an den Nobelpreis 2014 für Chemie. Nein? Der ging an zwei Amerikaner und einen Deutschen, William E. Morner, Eric Betzig und Stefan Hell für the development of super-resolved fluorescence microscopy, was übersetzt nicht anderes heißt als für „die Entwicklung hochaufgelöster Fluoreszenzmikroskopie“.
Es geht dabei immer um Mikroskopiemethoden, die mit Licht aus dem sichtbaren Teil des elektromagnetischen Spektrums funktionieren. Elektronenmikroskopie zählt nicht dazu, obwohl diese Technik die beste Auflösung hat. Allerdings ist die Elektronenmikroskopie auch recht beschränkt in dem was sie darstellen kann. Ein bisschen etwas darüber habe ich bereits unter Besuch bei Robert Mikroskopen aufgeschrieben.
Zurück zum sichtbaren Licht. Genau das stellt uns, durch seine Wellennatur, ein Hindernis in den Weg, wenn wir Mikroskopie machen wollen: Die Beugungsgrenze des Lichts. Dies allein ist schon eine spannende Geschichte für sich, und ein bisschen was davon habe ich bei Ernst Abbe war ein faszinierender Mensch aufgeschrieben. Für uns reicht es hier einfach zu wissen, das zwei Lichtpunkte nicht mehr von einander zu unterschieden sind, wenn sie zu eng zusammenstehen. Die Schwierigkeit besteht dann, wenn die gemessene Distanz der beiden Lichtpunkte zueinander unter der halben Wellenlänge der verwendeten Lichtquelle liegt; der sogenannten Beugungsgrenze.
Allerdings gibt es Möglichkeiten diese Beugungsgrenze zu umgehen. Aber wie immer im Leben: so etwas ist nicht umsonst. Man handelt sich andere Schwierigkeiten und Probleme ein, je nachdem welche Strategie man nutzt, um das Licht quasi auszutricksen. Für diese Strategien gibt es nicht wirklich treffende Oberkategorien – finde ich – daher hab ich meine eigene kleine Einteilung gewählt: mathematisch, physikalisch und chemisch.
Mathematische Überlistung
Das wohl prominenteste Beispiel hier wäre wohl die strukturierte Beleuchtung, auf Englisch Structured Illumination Micorscopy (SIM). Kurz und kompliziert gesagt: Durch Transformationen in den Frequenzraum von Bildern können Frequenzen unterhalb der Beugungsgrenze sichtbar gemacht werden. Kurz und einfach gesagt: Wir benutzen diese lustigen Muster in zwei überlappenden Geländern (Blick auf Autobahnbrücken z.B.) oder bei kleinkarierten Hemden im Fernsehen, um Dinge zu sehen die kleiner sind als die Beugungsgrenze. Diese Muster nennt man auch Moiré-Effekt.
Bei der strukturierten Beleuchtung regt man die Farbstoffe in einer Probe (siehe Das bringt Farbe ins Leben) mit einem engen Streifenmuster an, das sinusförmig ist. Dieses Muster verschiebt man einige Male senkrecht zur Streifenrichtung, dreht es um 60°, verschiebt wieder, dreht es ein letztes Mal um 60° und verschiebt wieder. Am Ende bekommt man so 15 Bilder, je fünf Bilder von kleinen Verschiebungen der jeweiligen Rotationen. Diese Bilder werden dann einer mathematischen Transformation in den Frequenzraum unterzogen, der sogenannten Fourier-Transformation.
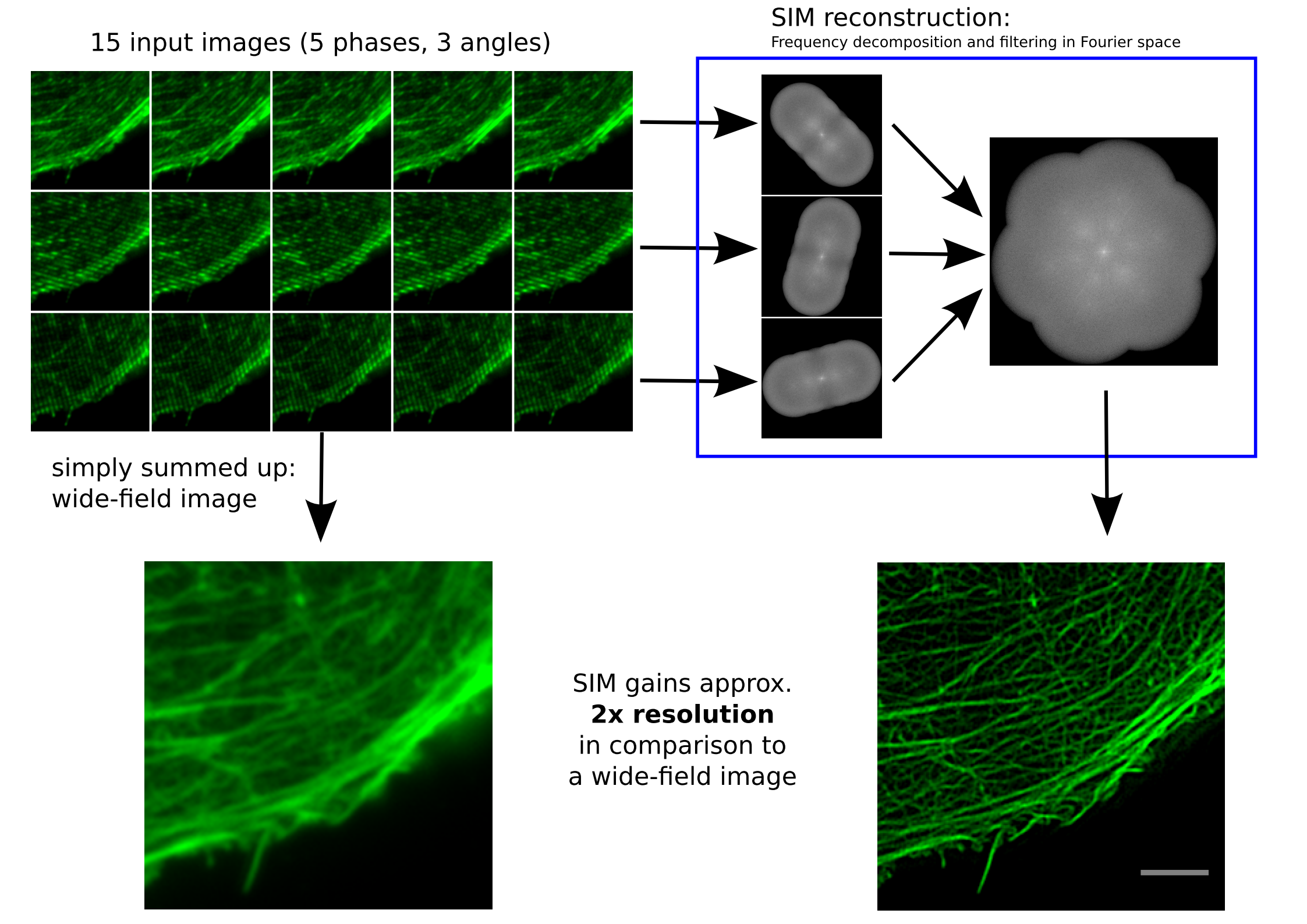
Funktionsweise SIM von Dr. Marcel Müller, Uni Bielefeld CC-BY-SA 4.0
Links, im Bild oben, sieht man 15 Bilder: die Rohdaten. Auf der rechten Seite kann man die Fouriertransformationen erkennen. Die drei „pillenfömigen“ Bilder sind die kombinierten Fouriertransformationen von den fünf Verschiebungen eines Winkels. In diesem Beispiel wurde Aktin angefärbt. In der Animation weiter unten, ist der eindimensionale Fall dargestellt. Die grauen Kästen stellen die markierten Strukturen in einer Probe dar, die blaue Linie ist das, was ein normales Fluoreszenzmikroskop sehen würde, die Sinusfunktion in türkis stellt die strukturierte Beleuchtung dar und in rot sind die Signale des SIM gezeigt. Man sieht, dass auf der linken Seite SIM einzelne Spitzen produziert, wohingegen im normalen Mikroskop (blau) keine getrennten Spitzen zu sehen wären.
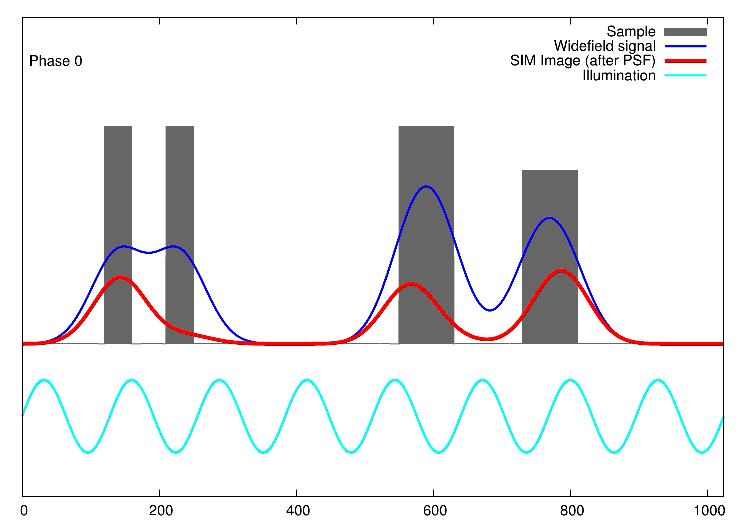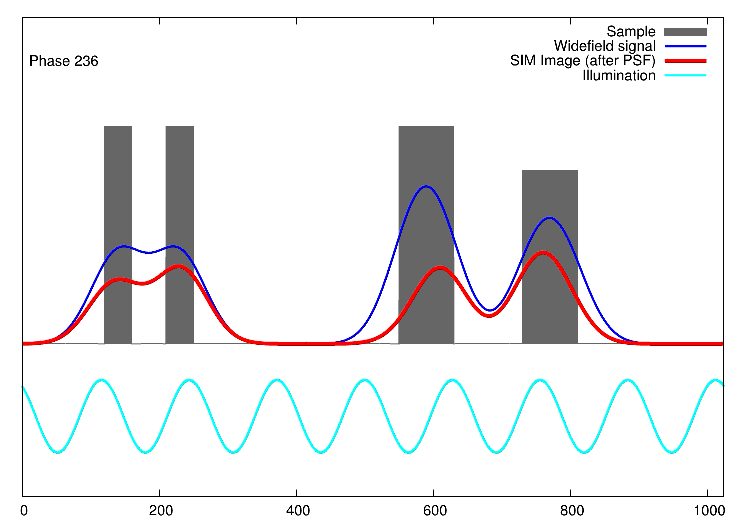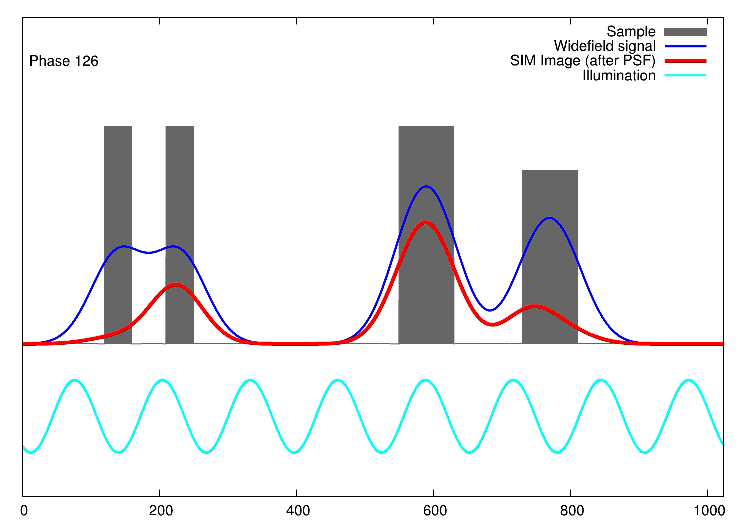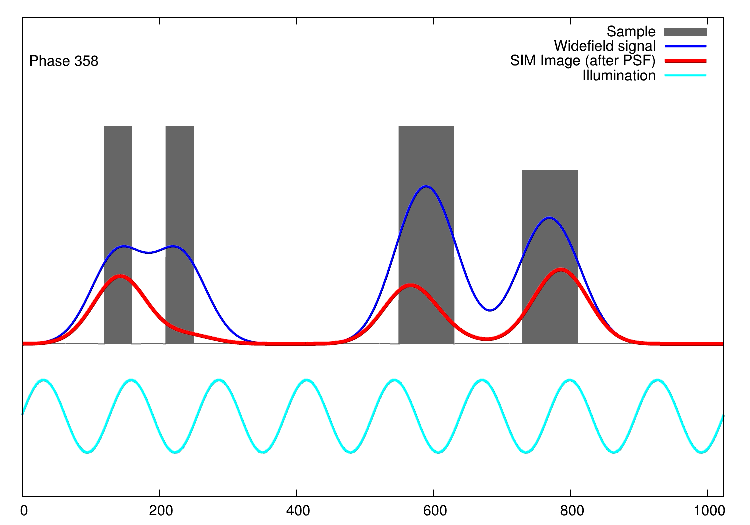
1D SIM von Dr. Marcel Müller, Uni Bielefeld CC-BY-SA 4.0
Diese Grafiken wurde von Marcel Müller an der Uni Bielefeld erstellt, der so etwas generell unter CC-BY-SA lizensiert. Nicht nur das möchte ich gerne erwähnt haben, sondern auch die Arbeit die Marcel in das Softwarepaket fairSIM gesteckt hat. Das ist ein offenes ImageJ Plug-In für SIM (Über ImageJ gibt es mehr bei Basteln ist auch Wissenschaft). Das ist deswegen so etwas Besonderes und Erwähnenswertes, weil es bisher so gut wie nur kommerzielle SIM-Software gegeben hat, von Mikroskopieherstellern die sich bei der Berechnung der Bilder nicht in die Karten schauen lassen wollten. Bei fairSIM kann man sich jede Zeile Programmcode anschauen, aber nicht nur das: fairSIM ist auch noch deutlich schneller als die meisten kommerziellen Programme und zur Zeit arbeiten die Forscher an der Uni Bielefeld an einer Variante die auf Grafikkarten noch ein Stück schneller rechnen können wird. So schnell, dass man höchstens einen Augenaufschlag auf das hochaufgelöste Bild warten muss, und so live seine Probe in SIM-Auflösung betrachten kann. Das ist eine Neuerung, die jeden Biologen begeistern wird und etwas, dass keine kommerzieller Hersteller anbietet. Yeah – open science!
Ich sprach darüber auch in meinem Vortrag auf dem 33. Chaos Communication Congress (33c3). Noch mehr Links zu SIM, Beispiele und den Vortrag selbst gibt es hier.
Stärken / Schwächen
Bei SIM ist eine recht aufwändige Nachbearbeitung der Messdaten durch einen Computer nötig. Dabei wird eine Auflösung von 100nm in der Ebene und 250nm in z-Richtung erreicht. Der große Vorteil ist hier, dass eigentlich alle gängigen Fluoreszenzfarbstoffe für die Technik geeignet sind, und es keinen Unterschied zu einer normalen Probenpräparation gibt.
Ein anderes Beispiel für eine mathematische Überlistung ist Super-resolution optical fluctuating imaging (SOFI).
Physikalische Überlistung
Man kann sich auch das Problem der Beugungsgrenze zu einem gewissen Grad ersparen, wenn man die beleuchtete Fläche kleiner macht. Zwar sind auch fokussierte Laser beugungsbegrenzt, können also das Kriterium „halbe Wellenlänge“ nicht unterschreiten, aber da geht es dann schon los mit der physikalischen Trickserei. Das wohl bekannteste Beispiel dafür wäre STED, Stimulated Emissioen Depletion oder, auf Deutsch stimulierte Emissionsverarmung. Dafür bekam Stefan Hell den Nobelpreis in Chemie 2014.
Dem STED-Ansatz zu Grunde liegt ein konfokales Mikroskop. Dabei wird ein Laser auf einen kleinen (beugungsbegrenzten) Punkt in der Probe foussiert, die Farbstoffe in diesem Punkt werde angeregt und die Fluoreszenz gemessen. Dann wird der Punkt ein kleines Stückchen in der Probe weiter bewegt. So kann man, Punkt für Punkt, Zeile für Zeile ein Bild einer Probe machen, und das geht auch schneller als es sich anhört. Oft braucht man für einen solchen Punkt in einem konfokalen Mikroskop höchstens ein paar Millisekunden Messzeit. Die Trickserei von STED ist jetzt, dass der Messpunkt effektiv kleiner gemacht wird. STED benutzt dafür einen zweiten Laser – der erste Laser den es beim konfokalen Mikroskop gibt ist ja weiterhin beugungsbegrenzt. Der zweite, auch sogenannte STED-Laser wird durch eine Phasenplatte wie ein kleiner Donut geformt, in dessen Mitte der erste Laser liegt.

By Marcel Lauterbach – Own work (Original text: Selbst erstellt), CC BY-SA 3.0, Link
Der STED-Laser liegt mit seiner Wellenlänge im gleichen spektralen Bereich, in dem das ausgesendete Licht des benutzten Farbstoffs liegt. Das führt dazu, dass sich angeregte Elektronen in den Farbstoffen denken „Hey, da leuchtet jemand, das wollen wir auch!“ und ihre Energie in Form von Licht der selben Wellenlänge des STED-Laser abgeben. Das nennt man stimulierte Emission. Mit dem STED-Laser räumt man alle angeregten Farbstoffe um das Zentrum dieses Donuts sofort wieder ab. Wenn man jetzt die Leistung des zweiten Laser erhöht, wird das Loch in der Mitte kleiner, und kann auch kleiner als die Beugungsgrenze werden. Deswegen habe ich oben von „effektiv kleiner“ gesprochen. So wird der Punkt, von dem man noch ein effektives Fluoreszenzsignal bekommt kleiner – und die Auflösung besser.
Stärken / Schwächen
Die erreichte Auflösung von STED liegt im Bereich von 50nm in der Ebene und mit mehr Trickserei bei 120nm in z-Richtung. Die Leistungsdichte durch die beiden Laser auf der Probe sind sehr hoch, wenn auch nur für kurze Zeit. Dies kann zum Bleichen der Farbstoffe und zur Beschädigung der biologischen Struktur selbst führen. Im Vergleich zu allen anderen Techniken ist das hochaufgelöste Bild direkt nach der Messung zu sehen, und keine Nachbearbeitung irgendwelcher Rohdaten im Computer ist nötig.
Ein anderes Beispiel für eine physikalische Überlistung ist das optische Rasternahfeldmikroskop (NSOM).
Photochemische Überlistung
Darunter fällt eine recht prominente Gruppe von Techniken, die einzemolekül lokalisationsbasierte Hochauflösungsmikroskopie, englisch singe molecule localizations.based super-resolution microscopy (SMLM). Für die Variante photo-activated localization microscopy (PALM) gab es für Eric Betzig und W. E. Moerner den Nobelpreis in Chemie 2014. Aber unter SMLM fallen noch andere Techniken, zum Beispiel direct stochastic optical reconstruction microscopy (dSTORM), die direkte stochastische optische Rekonstruktionsmikroskopie. Daran hab ich in meiner Doktorarbeit gearbeitet, und habe sie bei Ich hab was gegen Rauschen auch schon kurz erklärt – aber ich mache das hier noch mal etwas Ausführlicher und im Vergleich zu den anderen Techniken von oben.
Bei jeder SMLM Technik tauscht man den Raum gegen die Zeit. Das ist gar nicht esoterisch gemeint. Eine markierte biologische Struktur trägt, im Besten Fall, einen Haufen von Fluoreszenzfarbstoffen (siehe Das bringt Farbe ins Leben). Wenn ich jetzt dafür Sorge, dass die meisten dieser Farbstoffe kein Licht aussenden, am besten so viele, das man nur jeweils einzelne Moleküle sieht, dann kann man deren Ort recht genau bestimmen. In dem Fall hängt die Genauigkeit der Position nur von der Zahl der Photonen ab, die das Molekül aussendet und nicht mehr von der Beugungsgrenze. Die Signale sind immer noch beugungsbegrenzt, aber wenn man sicher sein kann, dass die Signale immer nur von einzelnen Molekülen ausgesendet werden, kann man auf die Position sehr genau zurück rechnen. Allerdings muss man jetzt verdammt viele Bilder machen, schließlich sollte jedes Molekül mindestens einmal dran sein mit Leuchten, mehrfach im Idealfall – sonst sieht man ja unter Umständen nicht alles von der biologischen Struktur, die von den Farbstoffen markiert wird. Im Falle von dSTORM sind das meistens so 10000 bis 20000 Bilder Rohdaten, aus denen man dann ein paar Millionen Koordinaten von Molekülen berechnet werden.
Die verschiedenen SMLM Techniken unterscheiden sich eigentlich nur über die Art und Weise wie diese langlebigen AUS-Zustände der Farbstoffe erreicht werden. Im Fall von dSTORM nutzen wir da einen besonderen Puffer, in den die Probe mit den Farbstoffen eingebettet ist. Der Puffer sorgt dafür, dass die Farbstoffe – bei recht starker Beleuchtung – ein große Chance haben in einen Radikalzustand reduziert zu werden. Dieser Zustand ist dunkel, was gut ist. Was nicht so gut ist: er ist auch chemisch recht aktiv und kann auch zur Zerstörung des Farbstoffes führen, aber irgendwas ist ja immer. Dieser dunkle Zustand ist aber auch langlebig wegen unserem Puffer. Wenn man sich an die Chemie in der Schule erinnert: Das Gegenteil von Reduktion ist die Oxidation – durch Sauerstoff würde der Farbstoff recht schnell aus dem dunklen Zustand zurück kommen. Unser Puffer bei dSTORM verhindert das, weil ein paar Enzyme enthalten sind, die den ganzen Sauerstoff schon in andere Verbindungen umgesetzt haben. Deswegen sorgt der Puffer dafür, dass der dunkle Zustand sehr langlebig ist und ein Großteil der Farbstoffe „aus“ ist.
Man nimmt sich also mehr Zeit, macht viele Bilder und bekommt am Ende viele Punkte aus denen man das Bild Punkt für Punkt wieder zusammen setzen kann. Ricardo Henriques, der eine Software Namens QuickPALM geschreiben hat, hat dazu ein ein schönes Video vom Eifelturm gebastelt, was diesen Ablauf verdeutlicht:
Ich sprach darüber auch in meinem Vortrag auf dem 33. Chaos Communication Congress (33c3). Noch mehr Links zu dSTORM, Beispiele und den Vortrag selbst gibt es hier.
Stärken / Schwächen
Die Rekonstruktion der vielen aufgezeichneten Bilder (10000-20000 bei dSTORM) geschieht nach der Datenaufnahme und benötigt einiges an Rechenleistung. Ein moderner Rechner ist damit schon einige Minuten beschäftigt. Die Auflösung von dSTORM kann bis zu 20nm in xy und 40nm in z erreichen. Einige Ansätze schaffen es sogar in den einstelligen Nanometerbereich, dabei handelt man sich aber deutlich längere Aufnahmezeiten und einige andere Nachteile ein – weswegen dafür auch nicht alle Proben geeignet sind. Der Ansatz, an dem ich in meiner Doktorarbeit gearbeitet habe, spectral demixing dSTORM (SD-dSTORM) erreicht 25nm in xy und 66nm in z, siehe dazu Ich hab was gegen Rauschen.
Wenn man Versuche an lebenden Zellen machen will oder Dynamiken beobachten möchte, ist das mit diesen Techniken, die die höchste Auflösung bieten, nur mit einem erheblichen Mehraufwand zu machen, und manchmal gar nicht.
Ich habe ja oben erwähnt der SMLM viele verschiedene Techniken enthält. Weitere Beispiele, neben dSTORM wären PALM oder uPaint.
Und wer gewinnt?
Das ist schwer zu sagen, welche Technik die Beste ist. Überall gibt es Vor- und Nachteile. Als kleine Einschätzungshilfe vielleicht: Mehr Auflösung bedeutet immer auch mehr Probleme. Wenn ich bei SIM bei der Probenpräparation kaum etwas anders machen muss im Vergleich zur normalen Fluoreszenzmikroskopie, bin ich bei STED auf lediglich ein paar Farbstoffe beschränkt, die die hohen Leistungsdichten aushalten können. Bei dSTORM habe ich auch nur ein paar Farbstoffe zur Auswahl, die das gewünschte Blinkverhalten zeigen und ich brauche einen besonderen Puffer. SIM kann man noch relativ schnell machen, bei STED und den SMLM Techniken dauert es schon ein bisschen, bis man ein Bild hat. Noch gar nicht gesprochen haben wir über die Größe der Farbstoffe selbst, oder die Biegsamkeit der Antikörper mit denen man Strukturen markiert. Es gibt noch sehr viele kleine Details die hier eine Rolle spielen, man kann diese Liste sehr lange fortsetzen.
Für mich haben SIM und SMLM Techniken die Nase vorn, weil sie Rohdaten generieren, die man später mit Software auswertet. Das ist auf den ersten Blick eigentlich ein Nachteil – aber ich würde sogar so weit gehen, dass hier die Zukunft der Hochauflösungsmikroskopie liegt. Gerade bei der Software machen immer mehr Forschergruppen ihren code offen, so das andere von neuen Erkenntnissen profitieren können. Also kann man nicht nur sehr einfach bessere Bilder durch die Arbeit anderer Forscher aufnehmen, durch neue Software kann man auch seine alten Rohdaten vermutlich besser Rekonstruieren und neue Dinge lernen. Aber nicht nur das: Wenn man diese Techniken als das Sammeln von Rohdaten begreift, dann kann man noch viel mehr daraus ziehen, und nicht nur Bilder rekonstruieren. Zum Beispiel ist unser Ansatz spectral demixing dSTORM so gebaut, dass im Prinzip die Farbinformation auch aus den Rohdaten gezogen wird. Wenn man das verinnerlicht, sind gerade diese Techniken, die sich auf spätere Auswertung mit (offener) Software verlassen, die Ansätze mit dem größten Potential neue Erkenntnisse in den Lebenswissenschaften zu liefern.

Es geht um Daten, nicht nur um Bilder – Rohdaten sind geil! CC-BY 4.0 André Lampe, aus meinem Vortrag beim 33c3.
 orcid.org/0000-0003-0750-4757
orcid.org/0000-0003-0750-4757
Kommentare (11)