
Wenn man so in der Gegen herum forscht, dann kommt man manchmal auch zu einem Ergebnis. Das ist mir jetzt auch passiert, endlich ist ein wissenschaftlicher Artikel veröffentlicht worden, in dessen Autorenliste ich an erster Stelle stehen darf.
Im Rahmen meiner Doktorarbeit arbeite ich an einem Hochauflösungsmikroskop, einem dSTORM, was kurz für direct STochastic Optical Reconstruction Microscope ist. Zusammen mit meinem Boss Jan Schmoranzer und Georgi Tadeus haben wir uns eine Methode überlegt, mit der man zwei oder mehr Farben in einem Hochauflösungsmikroskop realisieren kann, und ganz nebenbei auch noch ein paar Fehlerquellen bei der Bilddarstellung umgehen kann. Es geht dabei um Fluoreszenzmikroskopie, über die ich hier schon geschrieben habe. Die Dinge in der Zelle, die man sich anschauen will, markiert man dabei mit Farbstoffen, am liebsten mit zwei oder mehr Verschiedenen, um etwas über das Verhältnis zwischen den markierten Strukturen lernen zu können. Leider sind Fluoreszenzmikroskope nicht in der Lage beliebig stark zu vergrößern, man stößt an ein Limit, die Beugungsgrenze. Alles was kleiner als diese Grenze ist kann man mit einem normalen Fluoreszenzmikroskop nicht mehr sehen. Darüber habe ich schon im Artikel über Ernst Abbe berichtet, der genau auf diesem Gebiet geforscht hat.
Hochauflösung?
Es gibt aber Möglichkeiten um die Beugungsgrenze zu umgehen, und ich schreibe “umgehen”, weil das nie ohne Einschränkungen geht. Wenn man eine höhere Auflösung erreichen will, dann muss man an einer anderen Stelle immer deutliche Abstriche machen. Es gibt dazu mehrere Ansätze, zum Beispiel die strukturierte Beleuchtung (SIM: structured illumination microscopy), STED (stimulated emission depletion) oder photoaktivierte Lokalisationsmikroskopie (PALM: photoactivated localization microscopy). Für PALM und STED gab es 2014 den Nobelpreis in Chemie.
Die Technik PALM gehört zu einer größeren Gruppe von Methoden die Beugungsgrenze zu umgehen, nämlich zur Einzelmolekül Lokalisationsmikroskopie (SMLM: single molecule localization microscopy). Zu dieser Gruppe gehören auch STORM und dSTORM. Die grundsätzliche Idee dahinter ist, dass man die Farbstoffe blinken lässt, und zwar so, dass man immer nur Signale von einzelnen Farbstoff-Molekülen bekommt ohne das sich diese überlappen. Davon macht man dann viele, sehr viele Bilder, einige tausend oder sogar einige zehntausend. Diese Bilder werden dann mit mit einer bestimmten Software untersucht, die an jedes Signal eine Funktion versucht anzupassen, und so den genauen Ort des Moleküls zurückrechnet. Mit diesem Haufen an Positionen kann man dann, wie im Pointillitismus, ein Bild aus vielen Punkten aufbauen oder rekonstruieren. Diese Methode hat Ricardo Henriques, Autor der freien Software QuickPALM, in einem kurzen Video schön illustriert (unten). Die Art der Bildrekonstruktion haben die Techniken PALM, STORM und dSTORM gemeinsam – sie unterscheiden sich in der Art und Weise wie man die Farbstoffe dazu bringt so vereinzelt zu blinken.
Was ist SD-dSTORM?
Bei dSTORM bringt man die Farbstoffe auf chemischem Weg zum blinken. Eine sauerstofffreie, reduzierende Lösung wird benutzt, um die Zustände der Farbstoffe so zu beeinflussen, dass ein Großteil einfach nicht fluoresziert und nur einige wenige leuchten. Das funktioniert nur mit organischen Farbstoffen, die die schöne Eigenschaft besitzen in dieser Lösung nach kurzem leuchten wieder in den dunklen Zustand zurück zu kehren. Dann haben andere Farbstoffe die Chance zufällig (stochastisch, siehe Name) in den fluoreszierenden AN-Zustand zu kommen und auch ein bisschen Signal bei zu tragen.
Wir haben jetzt noch ein SD vor das dSTORM gesetzt, was kurz ist für Spectral Demixing. Das ist unser Ansatz um diese Technik mit mehreren Farben machen zu können. Benutzt man nämlich einfach rote und grüne Farbstoffe funktioniert das zwar auch, man handelt sich aber einen Fehler in der Position der Lokalisationen der beiden Farben ein. Grün und rot liegen einige hundert Nanometer in der Wellenlänge auseinander und die Brechung in den verwendeten Linsen ist Wellenlängenabhängig. Das kennt man ja von einem Prisma. Zwar sind moderne Optiken in Mikroskopen meistens achromatisch, also so weit korrigiert, dass das nichts mehr ausmachen sollte, aber in der Hochauflösungsmikrokopie, wo es um eine Genauigkeit von hundertstel Mikrometer geht, hilft einem so eine Korrektur nur bedingt. Im Fall von dSTORM kann man Positionen von einzelnen Farbstoffen bis auf 20 Nanometer genau lokalisieren und der Fehler durch den Wellenlängenunterschied (chromatische Abberation) hat auch mit korrigierten Optiken noch knapp 100 Nanometer. Es gibt schon Strategien wie man das am Computer nachträglich korrigieren kann, aber solche Programme lassen den Fehler lediglich auf 15 bis 20 Nanometer schrumpfen aber eben nie ganz verschwinden.
Unsere Idee war es dieses Problem komplett zu umgehen in dem wir zwei Farbstoff genommen haben die recht nahe beieinander liegen, sich aber in ihrem Spektrum ein bisschen unterscheiden. Wir haben zwei rote Farbstoffe verwendet, die man mit dem Auge so gut wie gar nicht unterscheiden kann. Dann haben wir einen Spiegel vor die Kamera gesetzt, der bis zu einer bestimmten Wellenlänge das Licht passieren lässt aber oberhalb dieser Wellenlänge anfängt das Licht zu reflektieren. So entstehen zwei Bilder unserer Probe, die wir nebeneinander auf einer Kamera abgebildet haben. So sollten also von jedem Farbstoff ein Licht-Punkt-Paar entstehen, auf jeder Seite der Kamera ein Licht-Punkt pro Blink-Ereignis. Man kann das im unteren Bild im Kasten a) sehen. Von diesen Bildern haben wir normalerweise zehn bis zwanzigtausend Bilder pro Probe aufgenommen und dann mit einer Software die Positionen der einzelnen Licht-Punkte errechnen lassen. Die Software die wir dafür verwendet haben heißt rapidSTORM und ist kostenlos und open source.
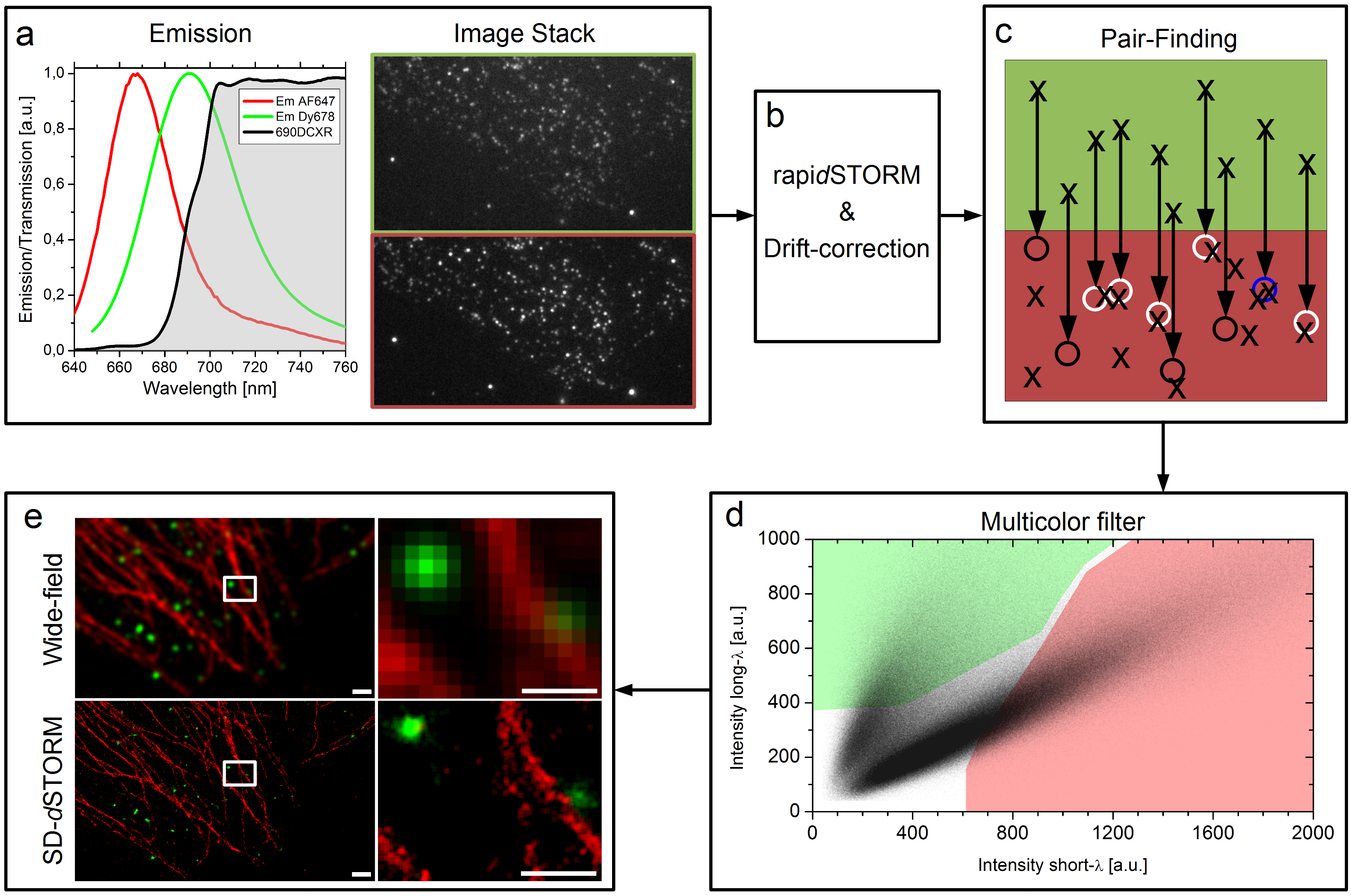
Der Workflow von SD-dSTORM. Lampe et al., Spectral demixing avoids registration errors and reduces noise in multicolor localization-based super-resolution microscopy, Methods and Applications in Fluorescence, Volume 3, Number 3, 13 August 2015 © IOP Publishing. Reproduced with permission. All rights reserved
Jetzt haben wir also einen Haufen von Punkten, oder besser gesagt haben wir Punkte-Paare, denn aus einem Blink-Ereigniss wurde durch den Spiegel vor der Kamera ein Licht-Punkte-Paar. Dieses Paar sagt uns direkt etwas über die Farbe, denn der hellrote Farbstoff (kürzere Wellenlänge) sollte auf der einen Seite einen helleren Lichtpunkt hinterlassen haben als auf der anderen, wohingegen der dunkelrote Farbstoff (längere Wellenlänge) sich genau anders herum verhalten sollte. Also müssen wir jetzt nachträglich aus der langen Liste von Positionen unsere Paare ausfindig machen. Das sieht man im Bild oben in Kasten c). Wie die Geometrie unserer Kamera und des Spiegels davor ist wissen wir, also wissen wir auch in welchem Abstand wir suchen müssen. Da aber keine Optik perfekt ist, suchen wir nach einem Partner in einer kleinen Umgebenung, was durch kleine Kreise am Ende der Pfeile angedeutet ist. Wenn da ein Blink-Ereignis im gleichen Bild statt gefunden hat, dann haben wir ein Paar gefunden, wenn wir kein Ereignis finden wird die ursprüngliche Position verworfen und wenn wir mehr als einen Partner finden können wir uns nicht mehr sicher über die Farbe sein und verwerfen alle beteiligten Lokalisationen. Das mag einem seltsam vorkommen, dass wir hier Daten “weg werfen”, aber das ganze hat auch einen Vorteil, wie man später noch sehen wird.
Am Ende haben wir also eine Liste von Paaren, immer jeweils zwei Lokalisationen, also auch zwei mal eine Helligkeitsinformation, einmal aus dem Kanal bei dem der Spiegel eine kürzere Wellenlänge durchgelassen hat und einem aus dem Kanal bei dem der Spiegel eine längere Wellenlänge reflektiert hat. Diese beiden Helligkeiten eines jeden Paars stellt man in einem Graphen da, wie man oben in Kasten d) sehen kann. Dort erkennt man schon zwei getrennte Populationen von Punktwolken, jeder Punkt steht für ein Paar. Und jetzt werden schon wieder Daten weggeworfen. Nur die Paare, die in einem der farbigen Bereiche liegen werden einem Farbstoff zugeordnet. Etwas irritierend hierbei unsere Farbenwahl: Der hellrote Farbstoff wird hier in rot dargestellt, der dunkelrote Farbstoff in grün. Längst nicht alle Paare liegen innerhalb der farbigen Flächen in d) und diese Paare werden auch weggeworfen. Wir machen das, weil man sich nicht überall in diesem Graphen sicher sein kann, dass dort definitiv nur Paare von dem einen Farbstoff gefunden werden und auf keinen Fall Paare von dem anderen. Das schlimmste was einem passieren kann ist einfach, dass man Lokalisationen eine falsche Farbe zuweist. Mit diesem “Multicolor filter” schaffen wir es die Falschzuweisung von Farben auf unter 1% zu drücken. Wie dann am Ende des rekonstruierte Bild aussieht, auch im Gegensatz zu einem Bild aus einem normalen Fluoreszenzmikroskop, sieht man oben in Kasten e). Warum es gar nicht verkehrt ist auch mal Daten weg zu werfen, sieht man im nächsten Bild.
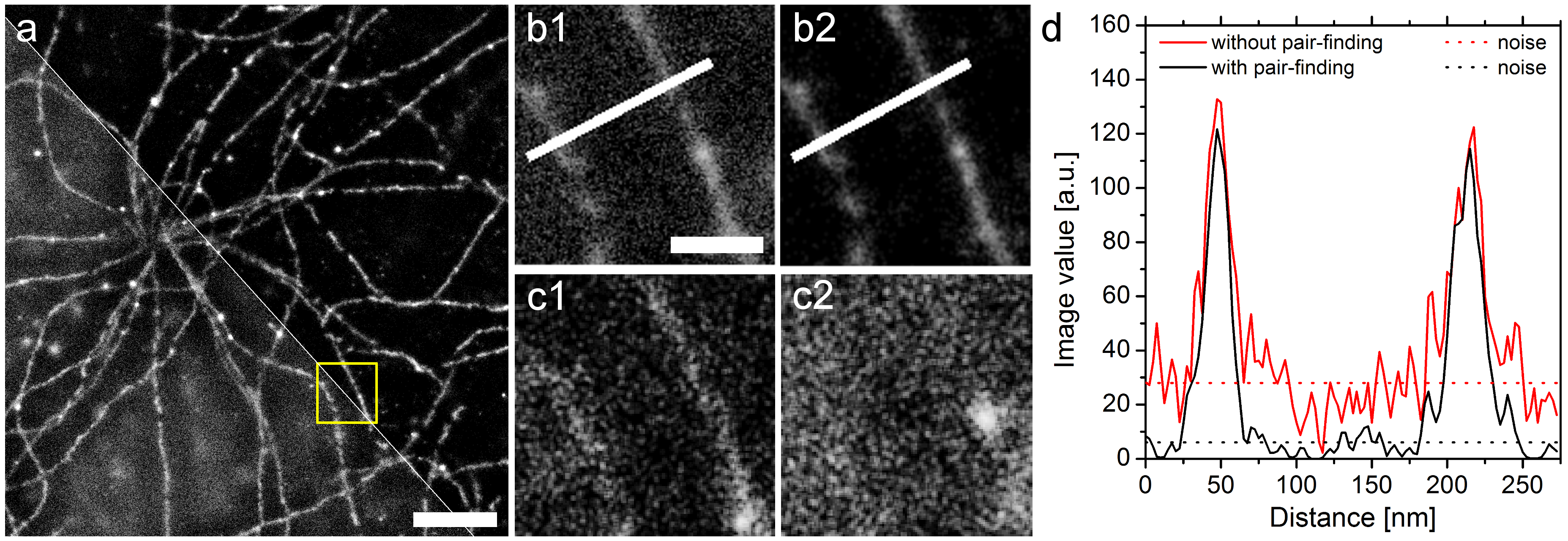
Besseres Signal-zu-Rauschen-Verhältnis mit SD-dSTORM. Lampe et al., Spectral demixing avoids registration errors and reduces noise in multicolor localization-based super-resolution microscopy, Methods and Applications in Fluorescence, Volume 3, Number 3, 13 August 2015 © IOP Publishing. Reproduced with permission. All rights reserved
Rauschen paart sich nicht
Bilder die man mit dem “normalen” dSTORM rekonstruiert sehen den Bildern unseres Ansatzes sehr ähnlich. Nur wenn man an einer schwierigen Stelle ein hoch-aufgelöstes Bild machen will, erkennt man dass das spectral demixing noch einen weiteren Vorteil hat. Wenn man bei einer Zelle direkt unter dem Zellkern ein Bild aufnimmt, dann befindet sich sehr viel Zelle über der Bild-Ebene und damit auch sehr viel Farbstoff an den markierten Strukturen außerhalb des Fokus unseres Mikroskops. Das führt dazu, dass die Software die nach Lichtpunkten sucht gar nicht so selten mal ein Blink-Ereignis findet, wo eigentlich keins gewesen ist. Wir nennen das dann auch einfach Rauschen oder noise, und das kann bei manchen Bildern schon recht störend sein, wenn man Details erkennen möchte. Im Bild über diesem Absatz kann man den Unterschied deutlich sehen. Dort haben wir ein einfarbiges Bild unterhalb des Zellkerns gemacht, weswegen man auch so eine spinnennetzartige Struktur sehen kann. Die Mikrotubuli, ein Teil des Skeletts der Zelle, die wir hier angefärbt haben entspringen nämlich direkt neben dem Zellkern. In den Vergrößerungen b1) und b2) sieht man den Unterschied zwischen dem normalen dSTORM und unserem SD-dSTORM, letzteres ist b2). Das wir wirklich nur Rauschen weggeworfen haben kann man in den Bildern c1) und c2) erkennen, dort haben wir Bildausschnitte der beiden Kameraseiten rekonstruiert die nur Lokalisationen zeigen, die von unserer Paar-Suche verworfen worden sind. Man kann zwar in c1) noch ein bisschen was von der Struktur erkennen, aber in c2) waren einfach keine Lokalisationen mehr übrig um diese Punkte zu verpaaren. Ein bisschen was haben wir also verloren, aber vor allem haben wir einen Großteil des Rauschens rausgeworfen. Der Graph in d) zeigt, dass das Signal kaum gelitten hat, das Rauschen aber deutlich weniger geworden ist.
Räumliches Sehen
Zum Schluss geht es um die dritte Dimension. Bereits in Ich sehe Batman (Lab Slang) hab ich einen Vesikel gezeigt, den wir mit SD-dSTORM in 3D abgebildet haben. Wie das funktioniert ist wirklich überraschend. Man muss dafür nämlich nicht mehrfach messen, sondern es reichen einem die zehn bis zwanzigtausend Bilder wie beim zweidimensionalen dSTORM. Es werden Bilder von einzelnen Blink-Ereignissen aufgezeichnet – wenn man jetzt aus diesen einzelnen Ereignissen nicht nur die x- und y-Koordinate berechnen könnte, sondern auch die Position in z, hätte man einfach so 3D-Informationen. Und in der Tat funktioniert es genau so. Es wird eine Zylinderlinse in den Strahlengang eingeführt, die jedes Blink-Ereignis verzerrt und zwar in Abhängigkeit zur Position des Farbstoffs in z. Liegt ein gerade blinkender Farbstoff in der Fokus-Ebene erhält man einen Punkt, liegt er oberhalb der Fokus-Ebene wird das Bild in x verzerrt, liegt er unterhalb wird das Bild in y verzerrt. Das funktioniert nur weil dSTORM so konzeptioniert wurde, dass die Farbstoffe immer einzeln liegen und sich die Signale untereinander nicht überdecken. Eine zweifarbige 3D Aufnahme ist im folgenden Bild zu sehen.
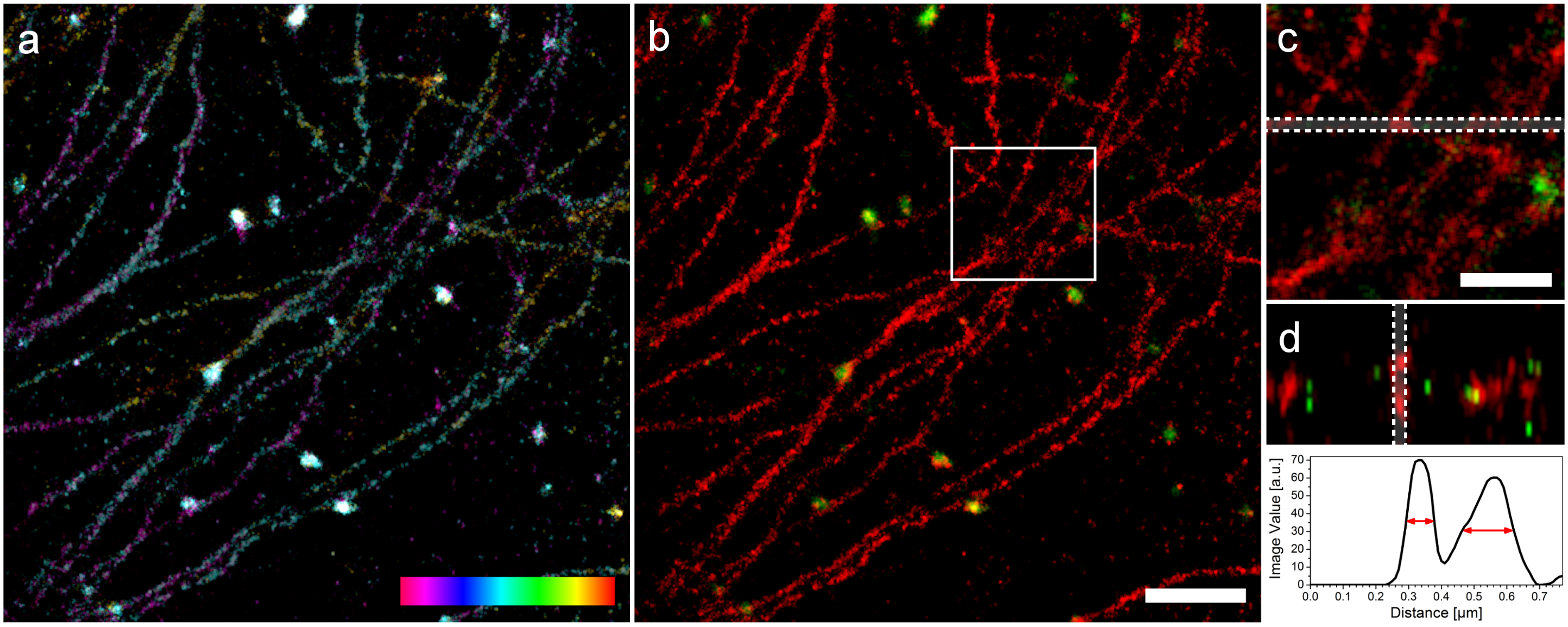
Zwei Farben 3D mit SD-dSTORM. Lampe et al., Spectral demixing avoids registration errors and reduces noise in multicolor localization-based super-resolution microscopy, Methods and Applications in Fluorescence, Volume 3, Number 3, 13 August 2015 © IOP Publishing. Reproduced with permission. All rights reserved
Oben sieht man Mikrotubuli und Vesikel. Die Mikrotubuli sind kleine Röhren mit einem Durchmesser von ungefähr 25nm und die Vesikel sind Kugeln mit einem Durchmesser von 150nm. Im Bild erscheinen diese Strukturen allerdings dicker, weil wir irgendwie den Farbstoff dran kleben müssen. Das geht über Antikörper, die leider auch eine eigene Größe haben und auf jede Struktur ungefähr 30nm draufschlagen (15nm auf jeder Seite). In a) sieht man zunächst die Struktur in 3D wobei die dritte Dimension farbcodiert ist, das linke, magentafarbene Ende liegt ganz unten, das rechte, rote Ende ganz oben und stellt einen Bereich von 200nm dar. Daneben sieht man das zwei Farben Bild und Detailansichten. In der ganz rechten, unteren Ecke sieht man die Bestimmung der Dicke von Mikrotubuli in z und wir sind recht stolz darauf. Da die Auflösung in z immer etwas schlechter ist, ist eine gemessene Dicke von knapp 100nm schon ein sehr gutes Ergebnis. Die Auflösung unseres SD-dSTORM beträgt in x und y 25nm und in z 66nm.
Bei diesen 3D Bildern sieht man schon, dass da deutlich mehr “gekrissel” zu sehen ist, also mittendrin auch mal Punkte die offenbar nicht zur Struktur gehören. Würde unser Ansatz nicht schon eine eingebaute Rauschreduktion besitzen, hätten wir diesen Spaß mit der dritten Dimension gar nicht treiben können, unser Signal wäre im Rauschen fast unter gegangen. Denn man bekommt kaum mehr Lokalisationen mit dem 3D Ansatz, man muss also mit fast genau so vielen Lokalisationen wie im 2D Fall gleich mehrere Bild-Ebenen mit kleinen Punkten ausstatten um etwas dreidimensionales zu rekonstruieren. Man könnte jetzt meinen, dass das nicht so richtig der große Wurf ist aber wissenschaftliche Veröffentlichungen, die zweifarbige 3D-Rekonstruktionen mit Hilfe von PLAM, STORM oder dSTORM zeigen, kann man an zwei Händen abzählen.
Unser Artikel ist beim Journal Methods and Application in Fluorescence und kann hier angeschaut werden. Die Software, die die Lokalisations-Paare sucht haben wir als open source veröffentlich. Sie heißt SDmixer und darüber haben wir auch einen Artikel beim selben Journal verfasst. Wer Lust hat mal mit dieser Software herum zu spielen, pre-build binaries für Windows 64bit gibt’s bei Sourceforge, mit Bedienungsanleitung und einem Beispieldatensatz. Den source code der Software kann man sich bei GitHub anschauen und wild für alle anderen Plattformen compilieren, wenn man Lust drauf hat.
 orcid.org/0000-0003-0750-4757
orcid.org/0000-0003-0750-4757
Kommentare (8)