
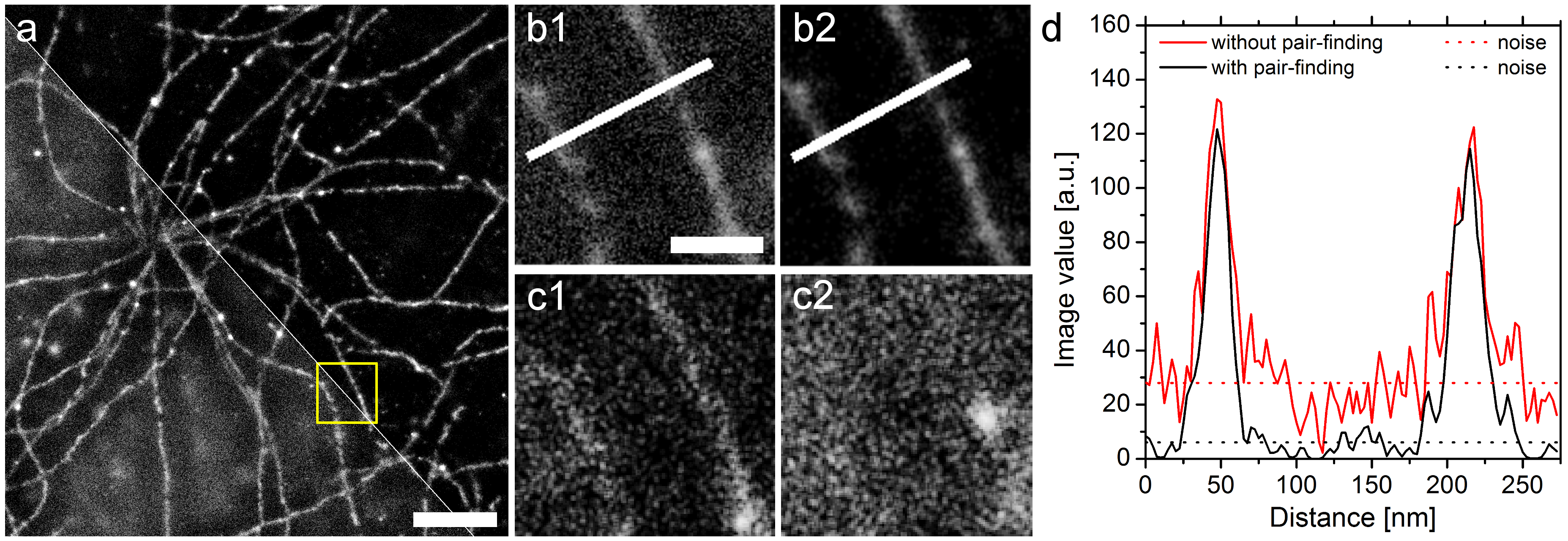
Besseres Signal-zu-Rauschen-Verhältnis mit SD-dSTORM. Lampe et al., Spectral demixing avoids registration errors and reduces noise in multicolor localization-based super-resolution microscopy, Methods and Applications in Fluorescence, Volume 3, Number 3, 13 August 2015 © IOP Publishing. Reproduced with permission. All rights reserved
Rauschen paart sich nicht
Bilder die man mit dem “normalen” dSTORM rekonstruiert sehen den Bildern unseres Ansatzes sehr ähnlich. Nur wenn man an einer schwierigen Stelle ein hoch-aufgelöstes Bild machen will, erkennt man dass das spectral demixing noch einen weiteren Vorteil hat. Wenn man bei einer Zelle direkt unter dem Zellkern ein Bild aufnimmt, dann befindet sich sehr viel Zelle über der Bild-Ebene und damit auch sehr viel Farbstoff an den markierten Strukturen außerhalb des Fokus unseres Mikroskops. Das führt dazu, dass die Software die nach Lichtpunkten sucht gar nicht so selten mal ein Blink-Ereignis findet, wo eigentlich keins gewesen ist. Wir nennen das dann auch einfach Rauschen oder noise, und das kann bei manchen Bildern schon recht störend sein, wenn man Details erkennen möchte. Im Bild über diesem Absatz kann man den Unterschied deutlich sehen. Dort haben wir ein einfarbiges Bild unterhalb des Zellkerns gemacht, weswegen man auch so eine spinnennetzartige Struktur sehen kann. Die Mikrotubuli, ein Teil des Skeletts der Zelle, die wir hier angefärbt haben entspringen nämlich direkt neben dem Zellkern. In den Vergrößerungen b1) und b2) sieht man den Unterschied zwischen dem normalen dSTORM und unserem SD-dSTORM, letzteres ist b2). Das wir wirklich nur Rauschen weggeworfen haben kann man in den Bildern c1) und c2) erkennen, dort haben wir Bildausschnitte der beiden Kameraseiten rekonstruiert die nur Lokalisationen zeigen, die von unserer Paar-Suche verworfen worden sind. Man kann zwar in c1) noch ein bisschen was von der Struktur erkennen, aber in c2) waren einfach keine Lokalisationen mehr übrig um diese Punkte zu verpaaren. Ein bisschen was haben wir also verloren, aber vor allem haben wir einen Großteil des Rauschens rausgeworfen. Der Graph in d) zeigt, dass das Signal kaum gelitten hat, das Rauschen aber deutlich weniger geworden ist.
Räumliches Sehen
Zum Schluss geht es um die dritte Dimension. Bereits in Ich sehe Batman (Lab Slang) hab ich einen Vesikel gezeigt, den wir mit SD-dSTORM in 3D abgebildet haben. Wie das funktioniert ist wirklich überraschend. Man muss dafür nämlich nicht mehrfach messen, sondern es reichen einem die zehn bis zwanzigtausend Bilder wie beim zweidimensionalen dSTORM. Es werden Bilder von einzelnen Blink-Ereignissen aufgezeichnet – wenn man jetzt aus diesen einzelnen Ereignissen nicht nur die x- und y-Koordinate berechnen könnte, sondern auch die Position in z, hätte man einfach so 3D-Informationen. Und in der Tat funktioniert es genau so. Es wird eine Zylinderlinse in den Strahlengang eingeführt, die jedes Blink-Ereignis verzerrt und zwar in Abhängigkeit zur Position des Farbstoffs in z. Liegt ein gerade blinkender Farbstoff in der Fokus-Ebene erhält man einen Punkt, liegt er oberhalb der Fokus-Ebene wird das Bild in x verzerrt, liegt er unterhalb wird das Bild in y verzerrt. Das funktioniert nur weil dSTORM so konzeptioniert wurde, dass die Farbstoffe immer einzeln liegen und sich die Signale untereinander nicht überdecken. Eine zweifarbige 3D Aufnahme ist im folgenden Bild zu sehen.
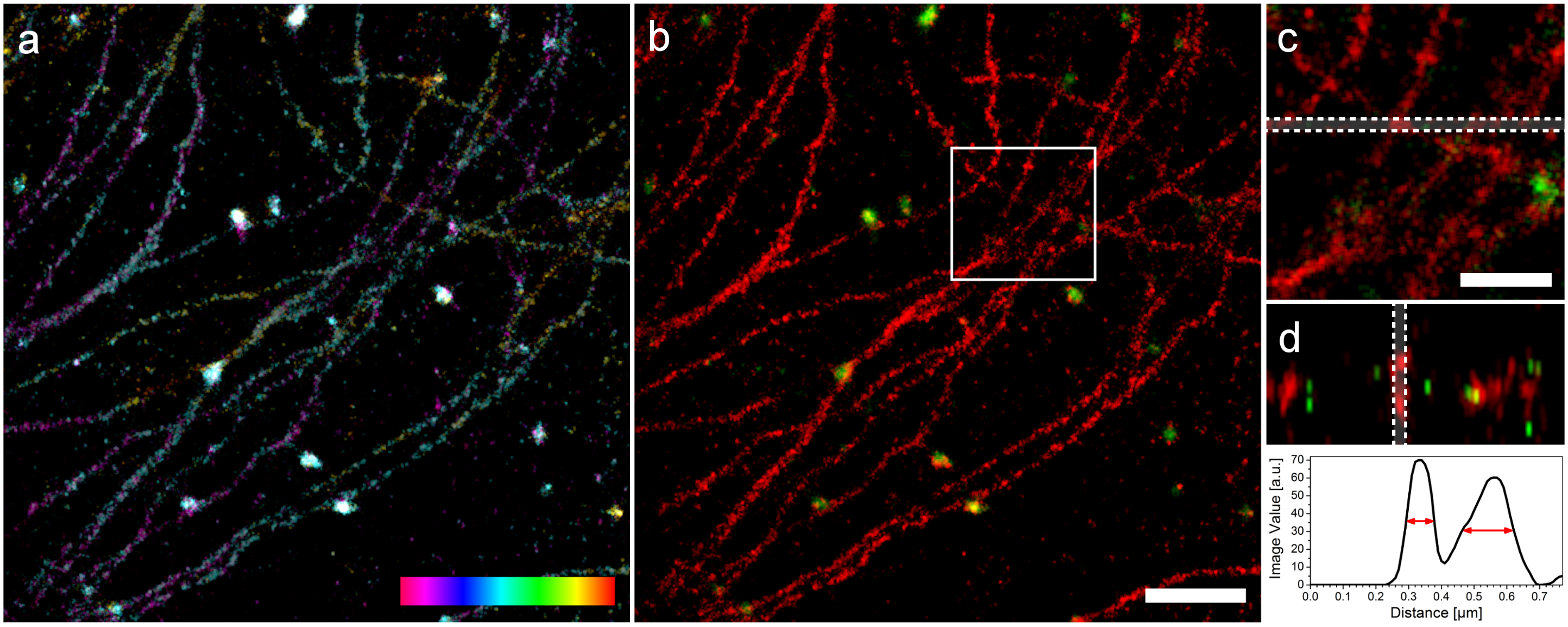
Zwei Farben 3D mit SD-dSTORM. Lampe et al., Spectral demixing avoids registration errors and reduces noise in multicolor localization-based super-resolution microscopy, Methods and Applications in Fluorescence, Volume 3, Number 3, 13 August 2015 © IOP Publishing. Reproduced with permission. All rights reserved
Oben sieht man Mikrotubuli und Vesikel. Die Mikrotubuli sind kleine Röhren mit einem Durchmesser von ungefähr 25nm und die Vesikel sind Kugeln mit einem Durchmesser von 150nm. Im Bild erscheinen diese Strukturen allerdings dicker, weil wir irgendwie den Farbstoff dran kleben müssen. Das geht über Antikörper, die leider auch eine eigene Größe haben und auf jede Struktur ungefähr 30nm draufschlagen (15nm auf jeder Seite). In a) sieht man zunächst die Struktur in 3D wobei die dritte Dimension farbcodiert ist, das linke, magentafarbene Ende liegt ganz unten, das rechte, rote Ende ganz oben und stellt einen Bereich von 200nm dar. Daneben sieht man das zwei Farben Bild und Detailansichten. In der ganz rechten, unteren Ecke sieht man die Bestimmung der Dicke von Mikrotubuli in z und wir sind recht stolz darauf. Da die Auflösung in z immer etwas schlechter ist, ist eine gemessene Dicke von knapp 100nm schon ein sehr gutes Ergebnis. Die Auflösung unseres SD-dSTORM beträgt in x und y 25nm und in z 66nm.
 orcid.org/0000-0003-0750-4757
orcid.org/0000-0003-0750-4757
Kommentare (8)