
Die DNA codiert ja bekanntlich die Erbinformation aller Lebewesen. Dazu verwendet sie “Worte”, die aus drei Buchstaben bestehen. Jedes dieser Worte (von speziellen Start/Stopp-Signalen mal abgesehen) steht dabei für eine Aminosäure. Da es 4 Buchstaben gibt, lassen sich 64 unterschiedliche Worte bilden. 4 Buchstaben sind natürlich ziemlich wenig. Könnten es auch mehr sein? Die Antwort auf diese Frage lautet: Vermutlich ja.
Die DNA codiert mit ihren 64 Worten insgesamt 20 unterschiedliche Aminosäuren – beispielsweise stehen die Worte GGT, GGC GGG und GGA alle für die Amonisäure Glycin. Oft liest man, dass es genau diese 20 Aminosäuren gibt, das ist aber eine Vereinfachung. Beispielsweise findet sich im Kollagen unseres Körpers häufig die Aminosäure Hydroxyprolin, für die es im genetischen Code gar kein Wort gibt – um Hydroxyprolin zu bekommen, wird die Aminosäure Prolin (mit dem genetischen Code CC-, der 3. Buchstabe ist beliebig) chemisch verändert. Insgesamt findet man in der Natur wesentlich mehr als nur 20 Aminosäuren – irgendwo habe ich mal die Zahl von 800 gelesen, habe aber gerade keine Quelle parat. Unser genetischer Code kann aber tatsächlich nur 20 von ihnen beschreiben.
Die einzelnen “Buchstaben” des genetischen Codes sind dabei Nukleinbasen, kleine Molekülbausteine, die in das Innere der Doppelhelix ragen, so dass sich jeweils zwei Buchstaben von jeder Seite der DNA zu einer “Leitersprosse. Dieses Bild hier zeigt die Struktur, die ja inzwsichen ohnehin vermutlich jeder kennt:
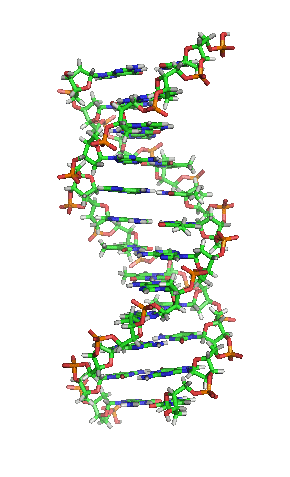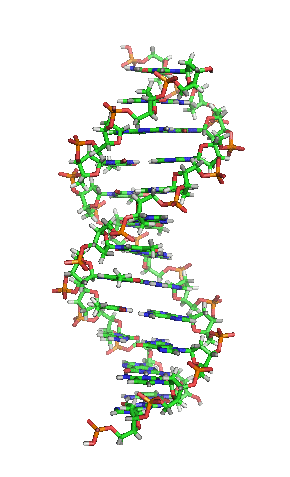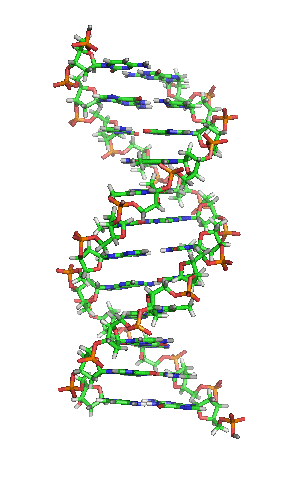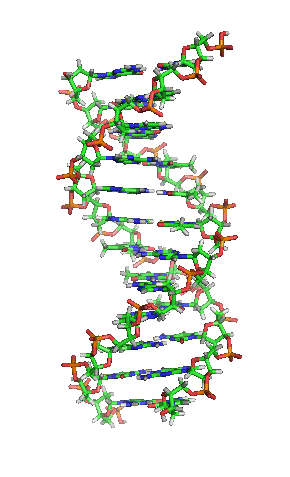
(Bild von Wikimedia, User Zephyris, CC License 3.0)
In der Mitte seht ihr, wie die Basen von den beiden Seiten zusammenpassen – chemische Bindungen (die berühmten Wasserstoffbrücken) sorgen dafür, dass die Basen aneinander gebunden sind und dass zu jeder der vier Basen genau eine andere passt.
Es ist schon seit längerem bekannt, dass ein DNA-Molekül auch andere Molekülgruppen in der Mitte besitzen könnte statt der vier Basen. Diese Molekülgruppen werden dabei nicht unbedingt durch Wasserstoffbrücken verbunden, sondern können auch einfach nur räumlich geeignet angeordnet sein, um zusammenzupassen und so die beiden Stränge zusammenzuhalten.
Aber funktioniert so etwas auch wirklich in einem Lebewesen? Um ein Lebewesen mit einer DNA mit künstlichen “Buchstaben” (in der Fachsprache heißen die UBPs – “unnatural base pairs”) auszustatten, genügt es ja nicht, einfach ein DNA-Stück mit einer solchen Struktur in das Lebewesen einzuschleusen. Zusätzlich muss die DNA ja auch bei der Zellteilung korrekt repliziert werden – die Zelle muss also in der Lage sein, auch die künstlichen Buchstaben beim Kopieren der DNA korrekt einzubauen.
Genau das ist vor kurzem gelungen. E. coli-Bakterien wurden mit Plasmiden, also kleinen DNA-Ringen, mit einigen UNBs versehen. Die beiden UNBs tragen dabei die klangvollen Namen d5SICS und dNaM – in der Arbeit werden sie gelegentlich aber auch einfach mit X und Y abgekürzt.So sehen die beiden Moleküle aus (oben im Bild, darunter zum Vergleich die beiden Buchstaben C und G mit ihren Wasserstoffbrücken; jeweils an der Seite seht ihr einen sechsfünfeckigen Ring, ein Zuckermolekül, das das “Rückgrat” des DNA-Strangs bildet):
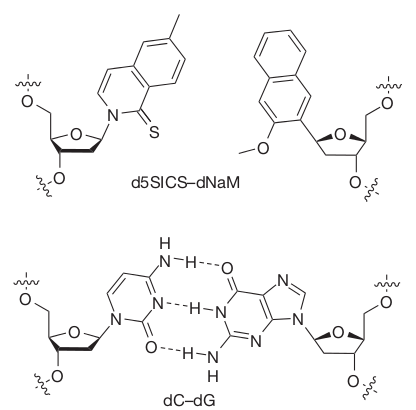
Aus Thyer&Ellefson, s.u.
Damit die Bakterien diese DNA auch vervielfältigen können, müssen sie natürlich die beiden Bausteine X und Y auch zur Verfügung haben. Hier steckte anscheinend eine der Hauptschwierigkeiten bei dieser Forschungsarbeit – da ich von Biochemie wenig Ahnung habe, tue ich lieber nicht mal so, als würde ich irgendwie verstehen, wie man nun genau die Enzyme ausgewählt hat, mit denen man die passenden Molekülbausteine X und Y in die Zellen bekommt und wie das im einzelnen funktioniert.
Auf jeden Fall hat man die E. coli-Bakterien so genmanipuliert, dass sie das passende Enzym herstellen können und dieses auch tatsächlich produzieren. Damit können sie also X und Y (beziehungsweise deren chemische Vorstufen, die noch eine Phosphor-Gruppe enthalten) aus ihrer Umwelt aufnehmen. (Auch hier gab es noch zusätzliche Probleme, weil die Vorstufen-Moleküle zu X und Y in einer Nährlösung nicht allzu stabil sind – man musste noch zusätzlich Kaliumphosphat zugeben, um das Zeugs zu stabilisieren. Vermutlich braucht man auch noch ein paar Billywig-Stachel…)
Die Buchstaben X und Y wurden in den Plasmiden auch nicht an irgendeine Stelle eingebaut, sondern dort, wo das Enzym DNA-Polymerase I für das Kopieren zuständig ist – dieses Molekül ist zumindest im Laborversuch in der Lage, DNA-Stücke mit den künstlichen Buchstaben zu kopieren.
Die so manipulierten Bakterien durften sich dann (in der geeigneten Nährlösung) vermehren, und tatsächlich zeigte sich, dass die DNA mit den zusätzlichen Buchstaben einigermaßen gut kopiert werden konnte – pro Kopierprozess gibt es eine Fehlerrate von etwa 0,6%, wenn ich alles richtig verstehe. Das ist immer noch viel, aber schon einigermaßen brauchbar. Interessant ist, dass anscheinend die Reparaturmechanismen für die DNA die künstlichen Buchstaben nicht eliminieren – sonst wäre das Experiment vermutlich gescheitert.
Mit X und Y stehen jetzt also zwei weitere Buchstaben im genetischen Code zur Verfügung. Leider tragen die Worte, die man mit diesen Buchstaben bilden kann, aber keinerlei Bedeutung: GCY codiert eben keine Aminosäure. Um die zusätzlichen Buchstaben auch nutzen zu können, muss man auch den Protein-Synthese-Apparat an die neuen Buchstaben anpassen. Sowohl das Ablesen der DNA (mit Hilfe der Boten-RNA) als auch das Übersetzen der Worte in Aminosäuren (mit Hilfe der Transfer-RNA) müssen an die neuen Buchstaben angepasst werden: Man müsste also insbesondere dafür sorgen, dass in den Zellen Transfer-RNA vorkommt, die an einem Ende das Wort GCY trägt und am anderen eine Aminosäure, die man in ein Protein einbauen möchte. Es ist also vermutlich noch ein weiter Weg hin zu Lebewesen, die die künstlichen Buchstaben nicht nur mit sich herumtragen, sondern auch nutzen können.
Auf jeden Fall zeigt die Forschung aber schon jetzt, dass es nicht absolut zwingend ist, dass die DNA genau die vier Buchstaben verwendet, die wir in der Natur vorfinden. Hätte die Evolution auch noch die Buchstaben X und Y ins Spiel gebracht, dann würde der genetische Code auch vielleicht Worte mit nur zwei Buchstaben verwenden – denn bei 6 Buchstaben sind das immer noch 36 mögliche Kombinationen. Ob eine solche alternative Evolution möglich wäre oder ob die anderen Buchstaben nicht doch irgendwelche Nachteile mit sich bringen, ist aber noch nicht klar.
Disclaimer Von Biochemie habe ich echt wenig Ahnung. Ich habe versucht, das wichtigste einigermaßen korrekt wiederzugeben, aber ich garantiere nicht dafür, dass ich alles richtig verstanden habe – erzählt das, was ich hier geschrieben habe, also nicht ohne weitere Kontrolle in irgendeiner Prüfung oder so…
ROSS THYER & JARED ELLEFSON
New letters for life’s alphabet
Nature vol 509 p. 291
Denis A. Malyshev, Kirandeep Dhami, Thomas Lavergne, Tingjian Chen, Nan Dai, Jeremy M. Foster, Ivan R. Correa Jr & Floyd E. Romesberg
A semi-synthetic organism with an expanded genetic alphabet
Nature vol 509 p. 385, doi:10.1038/nature13314





{kind=link}
Kommentare (38)