Zuvor müssen jedoch die DNA-Abschnitte, die die SNPs enthalten, aus der riesigen genomischen DNA aus der Speichelprobe erst einmal isoliert und vermehrt werden (der Großteil der genomischen DNA ist für diese Untersuchung irrelevant und würde nur stören). Dazu extrahiert man die DNA aus der Probe und reichert alle zu untersuchenden SNPs in einer großen Multiplex-PCR an. Die Primer für diese PCR sind so gewählt, daß die entstehenden PCR-Produkte die Regionen umfassen, in denen sich SNPs von Interesse befinden. In ihrer Sequenz entsprechen sie dabei den Primern, die auf dem Array aufgebracht sind.
Nach Abschluß der PCR werden die erzeugten Produkte noch chemisch fragmentiert, dadurch werden die die DNA-Stücke „handlicher“ und die Hybridisierung mit den Primern auf dem Array klappt besser. Von dieser Mischung fragmentierter PCR-Produkte wird dann etwas in einem Tropfen Flüssigkeit auf den Array aufgetragen. Daraufhin hybridisieren die DNA-Moleküle mit den auf dem Array fixierten Primern, so daß sich die letzte Base des Primers exakt an der letzten Base des DNA-Fragements vor dem SNP anlagert. Das Ganze passiert von beiden Seiten aus (also einmal „links“ und einmal „rechts“ vom SNP, das erhöht die Genauigkeit). Dann werden Polymerase und fluoreszenzmarkierte didesoxy(dd)-Nukleotide zugegeben. Diese ddNukleotide sind chemisch so verändert, daß eine Polymerase hinter ihnen kein weiteres Nukleotid in einen naszierenden DNA-Strang einbauen kann. Jedes der zugegebenen dd-Nukleotide, ddA, ddT, ddC und ddG, trägt dabei einen speziellen von vier verschiedenen Fluoreszenzfarbstoff (z.B. ddA immer gelb, ddT immer rot etc.), so daß man sie sozusagen an der Farbe unterscheiden kann.
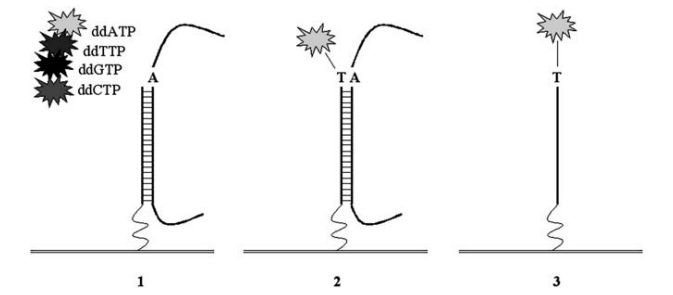
aus [1]

aus [2]
So wie auf dem Bild oben kann man dann sich einen solchen Array vorstellen: sehr viele verschiedene Primer sind eng an eng darauf befestigt. Dabei sind immer identische Primer in einem eng begrenzten Bereichd des Arrays („spot“) zusammen angeordnet, so wird eine Signalverstärkung erzielt.
Da die Position aller Primerspots auf dem Array bekannt ist, kann nun eine geeignete Kamera mit angepasster Bildauswertesoftware genau die Fluoreszenzsignale aller Spots auf dem Array auslesen, die durch Anregung der eingebauten Fluoreszenzfarbstoffe mit einem Laser induziert werden und aufgrund der Farbsignale auf die Identität der angefügten Nukleotide schließen. Ein gelber Spot etwa zeigt an, daß dort ein „A“ eingebaut wurde (und daß der Proband daher ein „T“-Allel in seinem SNP hat). Dabei sind natürlich auch Farbmischungen auswertbar, wie sie bei heterozygoten Genotypen (AT, gelb und rot) entstehen würden.
Nach Normalisierung und „Reinigung“ der Daten von Hintergrundrauschen und weiteren bioinformatischen Prozessierungsschritten gibt dann die Software für jeden SNP auf dem Array den Genotypen des Probanden aus. Das sind dann die Rohdaten des Tests, die ja auch Teil des Berichts waren. Hernach erfolgt noch die Interpretation der Ergebnisse auf Grundlage der Evidenz aus GWAS in Hinsicht auf die Einschätzung des genetischen Risikos des Probanden für die in die Untersuchung einbezogenen Krankheiten und auch diese Ergebnisse kommen in den Bericht.
Et voilà.
_____
Referenzen:
[1] Pullat, J., & Metspalu, A. (2008). Arrayed primer extension reaction for genotyping on oligonucleotide microarray. Prenatal Diagnosis, 161-167.
[2] P.J. Ulfendahl, Arrayed primer extension technique for nucleic acid analysis, Google Patents, 2001.
![]()

Kommentare (8)