Dieser Artikel ist als Anlage zu meinem Gentest-Selbstversuch-Artikel zu verstehen, sozusagen als “Supplementary Material” :-). Hierin erkläre ich, wie das Verfahren zur DNA-Analyse beim von mir ausprobierten Gentest technisch funktioniert. Weil das nicht für alle interessant (und auch nicht ganz unkompliziert) ist, habe ich das ganze hierhin ausgegliedert. Als Vorbereitung bzw. zusätzliche Lektüre empfehlen sich die Artikel zu DNA und PCR (ohne Kenntnisse über die Prinzipien der PCR ist der folgende Artikel eher unverständlich).
Die Firma futuragenetics benutzt das sogenannte APEX-Verfahren. Dieses Akronym steht für „arrayed primer extension“ (dt. Verlängerung mittels angeordneter Primer) und bezeichnet ein einfaches und robustes enzymatisches Verfahren zur gleichzeitigen Typisierung hunderter bis Tausender Variationen im Genom in einer einzigen Multiplex-Reaktion.
Die Gentests, wie auch der, den ich selbst ausprobiert habe, beruhen in der Regel darauf, daß der Status mehrerer SNPs erhoben wird. SNPs sind Polymorphismen, die jeweils nur eine einzige Base der DNA betreffen und fast immer liegt ein SNP in einer von zwei (ganz selten drei) Varianten oder „Allelen“ vor, z.B. A oder C. Jeder Mensch hat nun von jedem SNP zwei Kopien, eine auf jedem Partner eines Chromosomenpaars: eine Kopie kommt also vom Vater, die andere von der Mutter. So ergibt sich stets eine Kombination der Allele, die als „Genotyp“ bezeichnet wird. Von jedem SNP gibt es also drei mögliche Genotypen: zwei mal homozygot (z.B. AA und CC) und einmal heterozygot (AC).
Der SNP selbst hat übrigens in den wenigsten Fällen direkt mit der eigentlich untersuchten Prädisposition für eine Erkrankung zu tun (gibt es aber auch), oft liegt er nichtmal im kodierenden Bereich des Gens, das mit der Krankheit in Verbindung steht. Er liegt aber zumindest so nahe am für die Krankheit relevanten genetischen Bereich, daß er daran gekoppelt ist, also stets zusammen mit dem Gen vererbt wird. Das heißt für unser Beispiel, daß wenn das Allel „A“ unseres SNPs neben einer genetischen Variante liegt, die mit einem höheren Krankheitsrisiko verbunden ist, dann wird wegen der Kopplung das A-Allel auch immer mit dieser Genvariante zusammen vererbt. Findet man also in der DNA einer Person dieses A-Allel, so weiß man, daß in der Nähe auch die pathogene Genvariante liegt. Man spricht dann von Assoziation: es reicht daher aus, nur das Allel des naheliegenden, also assoziierten SNPs anzuschauen, um zu wissen, daß das untersuchte Individuum auch die entsprechende Genvariante trägt. Daß diese Zusammenhänge überhaupt existieren, muß zuerst in genomweiten Assoziationsstudien (GWAS) herausgefunden werden. Dann aber ist es sehr praktisch, weil sich durch Verfahren wie APEX SNPs sehr schnell, leicht und in hoher Multiplexität (= viele gleichzeitig) untersuchen lassen.
Das kann man sich – Achtung, Analogie – vielleicht so vorstellen, wie Barcodes an verschlossenen Koffern: die Barcodes sind fest mit dem Koffer verbundenund geben Auskunft darüber, was im Koffer drin ist. (Daß das so ist, hat man vorher herausgefunden, indem man 10.000 Koffer aufgemacht, den Inhalt mit dem Barcode abgeglichen und dabei gesehen hat, daß der xy-Barcode immer an Koffern mit schmutziger, der xx Barcode hingegen  immer an Koffern mit sauberer Wäsche klebt). Es reicht also, schnell den Barcode zu scannen, um den Inhalt des Koffers zu kennen, was viel leichter ist, als aufwendig das Kofferband zu entfernen, den Koffer aufzuschließen, zu öffnen und zu durchwühlen (und das will man vielleicht auch nicht unbedingt ;-).
immer an Koffern mit sauberer Wäsche klebt). Es reicht also, schnell den Barcode zu scannen, um den Inhalt des Koffers zu kennen, was viel leichter ist, als aufwendig das Kofferband zu entfernen, den Koffer aufzuschließen, zu öffnen und zu durchwühlen (und das will man vielleicht auch nicht unbedingt ;-).
Beim APEX-Verfahren werden nun Primer, also kurze Oligonukleotide, auf einer festen Unterlage fixiert, etwa einer Glasplatte, die man “Microarray” nennt. Die Sequenz dieser Primer ist genau komplementär zu der DNA-Sequenz, die exakt vor dem SNP, den man untersuchen will, liegt. Der SNP selber wird vom Primer jedoch nicht erfasst. Auf einem solchen Microarray haben zigtausende Primer Platz, die beliebige Sequenzen haben können, so daß mit einem einzigen Array viele Tausend SNPs gleichzeitig untersucht werden können.
Zuvor müssen jedoch die DNA-Abschnitte, die die SNPs enthalten, aus der riesigen genomischen DNA aus der Speichelprobe erst einmal isoliert und vermehrt werden (der Großteil der genomischen DNA ist für diese Untersuchung irrelevant und würde nur stören). Dazu extrahiert man die DNA aus der Probe und reichert alle zu untersuchenden SNPs in einer großen Multiplex-PCR an. Die Primer für diese PCR sind so gewählt, daß die entstehenden PCR-Produkte die Regionen umfassen, in denen sich SNPs von Interesse befinden. In ihrer Sequenz entsprechen sie dabei den Primern, die auf dem Array aufgebracht sind.
Nach Abschluß der PCR werden die erzeugten Produkte noch chemisch fragmentiert, dadurch werden die die DNA-Stücke „handlicher“ und die Hybridisierung mit den Primern auf dem Array klappt besser. Von dieser Mischung fragmentierter PCR-Produkte wird dann etwas in einem Tropfen Flüssigkeit auf den Array aufgetragen. Daraufhin hybridisieren die DNA-Moleküle mit den auf dem Array fixierten Primern, so daß sich die letzte Base des Primers exakt an der letzten Base des DNA-Fragements vor dem SNP anlagert. Das Ganze passiert von beiden Seiten aus (also einmal „links“ und einmal „rechts“ vom SNP, das erhöht die Genauigkeit). Dann werden Polymerase und fluoreszenzmarkierte didesoxy(dd)-Nukleotide zugegeben. Diese ddNukleotide sind chemisch so verändert, daß eine Polymerase hinter ihnen kein weiteres Nukleotid in einen naszierenden DNA-Strang einbauen kann. Jedes der zugegebenen dd-Nukleotide, ddA, ddT, ddC und ddG, trägt dabei einen speziellen von vier verschiedenen Fluoreszenzfarbstoff (z.B. ddA immer gelb, ddT immer rot etc.), so daß man sie sozusagen an der Farbe unterscheiden kann.
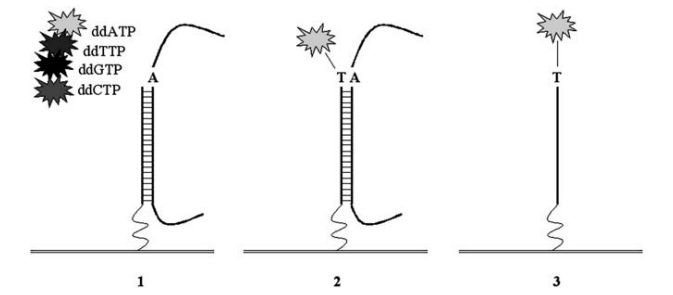
aus [1]

aus [2]
So wie auf dem Bild oben kann man dann sich einen solchen Array vorstellen: sehr viele verschiedene Primer sind eng an eng darauf befestigt. Dabei sind immer identische Primer in einem eng begrenzten Bereichd des Arrays („spot“) zusammen angeordnet, so wird eine Signalverstärkung erzielt.
Da die Position aller Primerspots auf dem Array bekannt ist, kann nun eine geeignete Kamera mit angepasster Bildauswertesoftware genau die Fluoreszenzsignale aller Spots auf dem Array auslesen, die durch Anregung der eingebauten Fluoreszenzfarbstoffe mit einem Laser induziert werden und aufgrund der Farbsignale auf die Identität der angefügten Nukleotide schließen. Ein gelber Spot etwa zeigt an, daß dort ein „A“ eingebaut wurde (und daß der Proband daher ein „T“-Allel in seinem SNP hat). Dabei sind natürlich auch Farbmischungen auswertbar, wie sie bei heterozygoten Genotypen (AT, gelb und rot) entstehen würden.
Nach Normalisierung und „Reinigung“ der Daten von Hintergrundrauschen und weiteren bioinformatischen Prozessierungsschritten gibt dann die Software für jeden SNP auf dem Array den Genotypen des Probanden aus. Das sind dann die Rohdaten des Tests, die ja auch Teil des Berichts waren. Hernach erfolgt noch die Interpretation der Ergebnisse auf Grundlage der Evidenz aus GWAS in Hinsicht auf die Einschätzung des genetischen Risikos des Probanden für die in die Untersuchung einbezogenen Krankheiten und auch diese Ergebnisse kommen in den Bericht.
Et voilà.
_____
Referenzen:
[1] Pullat, J., & Metspalu, A. (2008). Arrayed primer extension reaction for genotyping on oligonucleotide microarray. Prenatal Diagnosis, 161-167.
[2] P.J. Ulfendahl, Arrayed primer extension technique for nucleic acid analysis, Google Patents, 2001.
![]()

Kommentare (8)