Ende 2019 ist in Deutschland die Strafprozessordnung aktualisiert worden. In einem Bericht darüber schrieb ich:
Die Änderung der StPO sieht vor, daß künftig auch Haar-, Augen- und Hautfarbe sowie das Alter einer tatverdächtigen Person im Rahmen einer strafrechtlichen Ermittlung aus der DNA ausgelesen werden dürfen.
Man bezeichnet diese Untersuchungen auch als forensische DNA-Phänotypisierung. Kurz nach der Anpassung der StPO an die Moderne hatten wir damals noch in Kiel damit begonnen, eine Methode zur Vorhersage der Haut-, Haar- und Augenfarbe zu etablieren und inzwischen läuft sie dort ganz gut. Auch hier in meiner (nicht mehr so ganz) neuen Abteilung in Köln bieten wir diese Untersuchungsform an. Die molekulargenetische Grundlage besteht (wie unter obigem Link nachzulesen) in der Typisierung verschiedener, mit den möglichen Ausprägungen der oben genannten äußerlich sichtbaren Eigenschaften assoziierter Einzelnukleotidpolymorphismen (SNP). Die Typisierungsdaten werden dann mit einem speziellem Alogrithmus (z.B. diesem) ausgewertet. Ausführlicher ist das in meinem Artikel von 2014 noch einmal nachzulesen.
Aber was ist mit dem Alter? In vielen kriminalistischen Lagen kann es zwischen sehr interessant bis entscheidend sein, das Alter einer tatbeteiligten Person zu kennen, z.B. um bestimmte Personengruppen als Täter auszuschließen oder um Ermittlungen zu konzentrieren und zu priorisieren.
Die Bestimmung des Alters aus der DNA wird ja ausdrücklich auch in der StPO erlaubt,
Ist unbekannt, von welcher Person das Spurenmaterial stammt, dürfen zusätzlich Feststellungen über die Augen-, Haar- und Hautfarbe sowie das Alter der Person getroffen werden. (StPO, §81e, Molekulargenetische Untersuchungen)
doch das Lebensalter einer Person kann nicht durch SNP-Analyse abgeschätzt werden, da sich deren Konfiguration mit fortschreitender Zeit nicht ändert. Stattdessen schaut man sich zur Schätzung des Alters einer Person das Methylierungsmuster ihrer DNA an und weil wir hier in Köln auch diese Methode bereits in der Fallarbeit einsetzen, möchte ich heute mal darüber berichten, wie das eigentlich funktioniert (die klassischen, anthropologischen bzw. bildgebenden Methoden der Altersschätzung sind hier nicht Gegenstand der Betrachtung).
Hinweis: Um den Artikel besser nachvollziehen zu können, z.B., wenn man gerade über das Wort “Methylierungsmuster” gestolpert ist, empfehle ich sehr die vorherige Lektüre dieses Basics-Artikels.
Die Voraussetzung für die molekulargenetische Altersschätzung ist logischerweise irgendeine meßbare Struktur, Muster, Vorgang etc., die sich mit dem biologischen Alter einer Person (das bei den meisten Menschen mehr oder weniger mit dem chronologischen Alter übereinkommt) in einem systematischen Verhältnis befindet, so, daß man von der Messung des letzteren und bei Kenntnis des mathematisch darstellbaren Verhältnisses ersteres berechnen oder besser schätzen kann, was dann einen Rückschluß auf das chronologische Alter zuläßt.
Was aber heißt hier “meßbar”? Wie mißt man die Methylierung von DNA? Es gibt dafür unterschiedliche Methoden, die verbreitetsten sind die differentielle enzymatische Spaltung von DNA mit methylierungssensitiven Restriktionsendonuleasen (MSRE) gefolgt von PCR, Affinitiäts-Capturing von methylierter DNA und die Natriumbisulfit-Konversions-Sequenzierzung. Nur die letztgenannte werden wir uns hier näher ansehen, denn sie erlaubt eine quantitative Bestimmung des Methylierungsgrads von DNA-Stellen (CpG-Inseln) und ist auch die Methode, die wir selbst anwenden.
Die Bisulfit-Konversion beruht darauf, daß sie nicht-methylierte Cytosin-Basen in der DNA in Uracil umwandelt, welches statt zu G(uanin) wie Cytosin zu A(denin) wie Thymin komplementär ist.
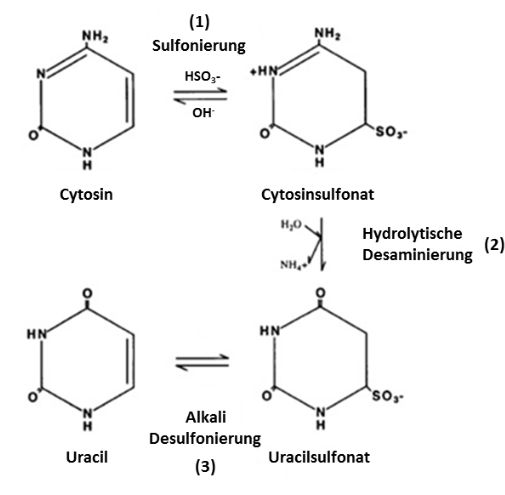
für die Sulfonierung (1) wird das Bisulfit (HSO3-) benötigt
Methylierte Cytosine hingegen werden nicht in Uracil umgewandelt, die angehängte Methylgruppe (CH3) schützt sie gewissermaßen vor dem Bisulfit. Nach einer vollständigen Konversion sind also alle nicht-methylierten Cytosine in einem DNA-Molekül zu Uracil umgewandelt worden. Wird nun von der DNA eine Kopie, wie z.B. in einer PCR, gefertigt, werden im komplementären Strang gegenüber den Uracilen Adenine eingebaut, die Basensequenz der Kopien verändert sich also gegenüber dem Original.
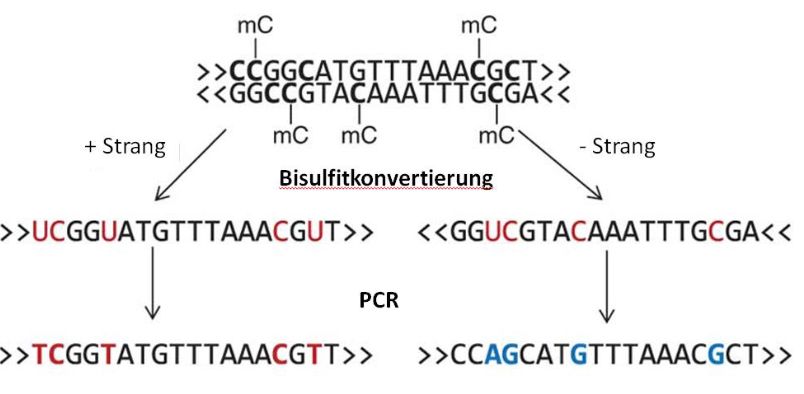
mC: Methylgruppe
Anschließend werden die kopierten DNA-Fragmente sequenziert, häufig wird dafür die sogenannte „Pyrosequenzierung“, bei der es sich um eine Variante des NGS handelt (und auf die ich hier nicht im Detail eingehen kann, sonst wird es zu kompliziert), eingesetzt. So kann man die Stellen, an denen sich methylierte Cytosine befunden haben, durch Vergleich mit einer Referenzsequenz (bei der Cytosine statt Thyminen stehen) erkennen und zusätzlich genau messen, zu welchem Grad, zu wieviel % eine Stelle im Genom methyliert ist (es ist nicht in allen Zellen die DNA identisch methyliert, wenn z.B. in der Hälfte der in einer Probe enthaltenen Zellen eine bestimmte CpG-Stelle in der DNA methyliert ist, dann wäre ihr Methylierungsgrad 50%).
![]()

Kommentare (6)