
UAP – die “Universal Analysis Pipeline” wurde bereits im letzten Jahr als “Pipeline”-Lösung für Bioinformatik-workflows publiziert[Kämpf et al., 2019]. Für diesen Artikel ist wichtig zu verstehen, dass genomorientierte Bioinformatik (wie auch nahezu alle komplexe Datenanalytik) nahezu jederzeit bedeutet eine Analyse in eine Vielzahl von Schritten (z. B. – Achtung starke Vereinfachung – Qualitätsbestimmung, Qualitätskontrolle, Mapping, Finden von Mutationen) aufzuteilen. Also bedarf es Lösungen, Software, wie sie UAP verspricht (hier aus dem abstract der Veröffentlichung, Anmerkung durch mich):
uap is a freely available tool that enables researchers to easily adhere to reproducible research principles for HTS (Anm: High throughput sequencing) data analyses.
Üblicherweise wird so eine mehrschrittige Analyse “Pipeline” genannt und die Systeme, welcher der Aufgabe nachkommen Analysen generisch zu verwalten und auszuführen sind sogenannte wissenschaftliche Workflow-Systeme. Über die Unterschiede und Hintergründe plane ich eine Miniserie, deshalb erspare ich Euch an dieser Stelle die Details …
Und UAP?
Workflow management systems are indispensable for reproducibly controlling more complex analyses, like HTS data analysis. Published workflow management systems for HTS data analysis are either highly flexible and made for experienced programmers or lack a lot of flexibility but can be used intuitively and some are specific to a certain type of analysis. In the introduction we listed four minimal requirements that we consider essential for ensuring reproducibility and consistency of HTS data analyses. Additionally, but beyond the programmatic control of a of a WMS, using versioned external data like genome sequences or annotation is key for reproducible analysis. None of the published systems we are aware of, however, completely satisfies these criteria. uap has been designed to fulfill these.
Klingt toll – genau das, worauf die Bioinformatik gewartet hat. Und gleich besser als vergleichbare Systeme! Und dies ist auch der Grund, warum diese Serie mal wieder eine bestimmte Software aufgreift.
Nun, ich habe am 8. Februar 2019 mal nachgefragt:
Dear uap developers,
I am interested in workflow management systems and in the comparison thereof. May I ask:
Is the development of uap ongoing?
This repo is given as the master repo in the uap paper. However, it is a fork from https://github.com/kmpf/uap . Can you elaborate on this?
Und die Antwort war, u.a.:
Yes, the development is ongoing, but the resuts are not displayed on github as of now.
Unterschiedliche verlinkte Quellen zum Download finde ich etwas unhandlich: Woher sollen interessierte Anwender wissen, was im Lauf der Zeit Bestand hat? Und muss man dann immer checken, was gerade aktuell ist? Im Allgemeinen ist ein Institutsaccount leichter zu pflegen als ein persönlicher Account wissenschaftlicher Wanderarbeiter (DoktorandIn oder PostDoc) – aber das ist bloß meine Meinung.
Jedenfalls zunächst für sich selbst zu entwickeln ist ganz normal – nur nach erfolgreicher Publikation (also dem Versprechen des “funktioniert wie beschrieben: Kollegen, ihr könnt das ausprobieren”) zeigt die öffentliche Weiterentwicklung an, dass das Projekt noch lebt. Dann sollte man doch öffentlich weiterentwickeln? Woher wissen potentielle Nutzer, ob das Projekt lebt? In den letzten Monaten kam wenig Code hinzu, aber seht selbst (Screenshot vom 22. Aug.) …
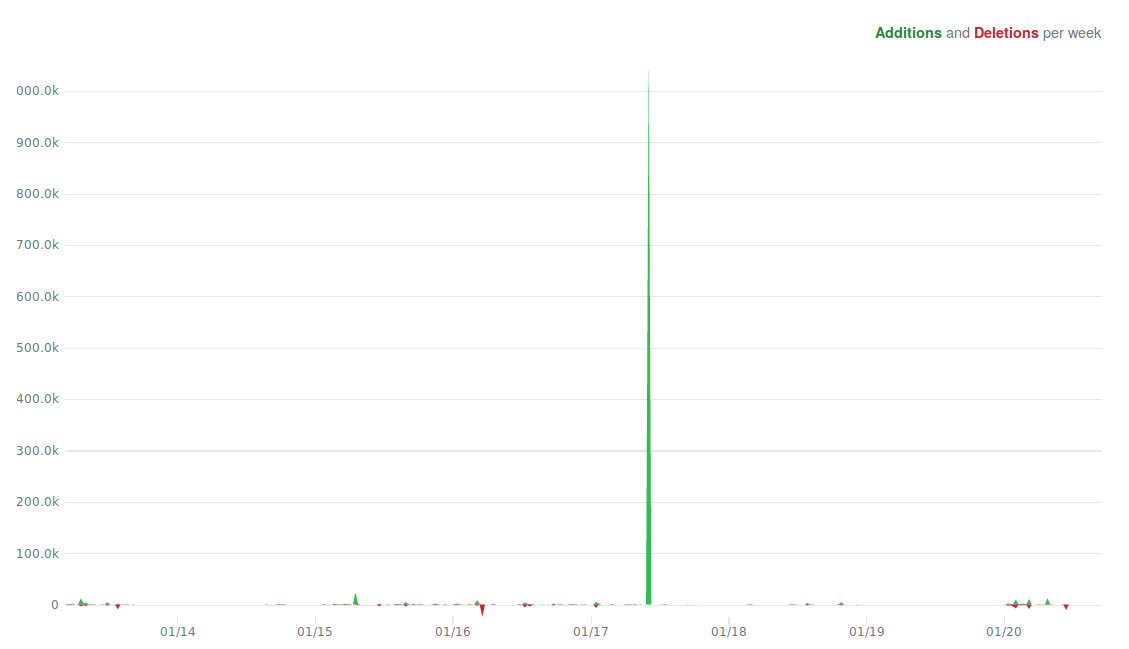
Frequenz der Änderungen gemäß github (link siehe Text). Grün: Hinzugefügter Code. Rot: Gelöschter Code. Das Changelog zeigt außerdem: Seit Veröffentlichung kam weder ein Release noch wesentlich Programmcode hinzu. Die meisten Änderungen beziehen sich auf die Dokumentation und die Code-Qualität.
Wie steht es um die Qualität?
Die Behauptung der UAP-Entwickler ist es gut zu sein, besser teilweise als andere Systeme in der eigenen Bewertung:

Vergleichsmatrix aus der Veröffentlichung. Linke Spalte die Eigenschaften, die Workflow-Systeme aufweisen sollten und dann in jeder weiteren Spalte Workflow-Systeme im Vergleich mit UAP. (Ausgefüllter Kreis: Feature vorhanden, weißer Kreis: nicht vorhanden, halb ausgefüllter Kreis: teilweise vorhanden).
In den Vergleich der Entwickler wurden diejenigen Systeme einbezogen, die ein seit nunmehr sechs Jahren nicht mehr gepflegtes Vergleichsprojekt hat, das Workflowsysteme aufführt, die aktiv von mehr als fünf EntwicklerInnen weiterentwickelt werden. Mich wundert etwas, warum snakemake[Köster and Rahmann; 2012] nicht aufgeführt wird: Neben Nextflow[Di Tommaso et al.; 2017] hat es nämlich auch HPC im Blick, unterstützt weitere Plattformen und hat eine relativ große Community – ist also in puncto Portierbarkeit ein ernsthafter Konkurrent . Auch ist es ebenfalls in Python geschrieben (was direkte Vergleichbarkeit der Codemetrik ermöglicht). Auch unterstützt snakemake ein Datenmanagementsystem (in der Vergleich gar nicht aufgeführt), ebenso auch Cloud-Umgebungen (auch nicht aufgeführt).
Ein weiterer Vergleich sei also erlaubt: uap hat insgesamt 11300 Codezeilen in Version 1.0. snakemake aktuell über 18.000. Das sagt nicht viel, aber Features ohne Code gibt es nicht. Bei uap sind sämtliche unterstützten Applikationen mit an Bord. Bei snakemake nicht, da gibt es ein Extra-Projekt mit separatem Code, bzw. Dokumentation. Die Aussage, dass uap bei weitem nicht so viele mögliche Workflows unterstützt, wie das ältere snakemake Projekt oder andere Workflow-Systeme, z. B. nextflow, ist durch die Links klar.Graphviz-2.42.2-foss-2019b-Python-3.7.4.eb
Wie auch immer – so eine Vergleichsmatrix ist absolut sinnlos, wenn man den Ist-Zustand der eigenen Software mit Publikationsbeschreibungen aktiv weiter entwickelter Software vergleicht – schließlich kann in 7-8 Jahren Softwareentwicklung ein Bisschen was passieren … So unterstützen andere Lösungen (z. B. wieder mal snakemake) weit mehr Plattformen als in der Vergleichsmatrix aufgeführt sind. Im Grunde wäre von mir unfair diese Matrix zu zeigen – wenn es ein neues Release gegeben hätte.
Warum ich überhaupt auf uap aufmerksam wurde ist aber das Versprechen Cluster zu unterstützen. Wie es damit ausschaut kann ich nicht sagen, in Version 1 baut die Dokumentation nicht, was mir erst beim Schreiben des Artikels aufgefallen ist. Eine Software, die nicht unmittelbar baut, weil die Installationsroutine einen Fehler aufweist, macht offengestanden wenig Spaß. Dokumentation sollte dabei sein – wenn man sich diese erst bauen muss, dann lieber auch ohne Probleme. Auch hier habe ich mir erlaubt mein Problem kund zu tun. Jedenfalls ist der Code zum Cluster-Support nichtssagend. Für den Cluster der Entwickler, mag uap daher hinreichend sein – für andere Cluster sicher nicht: Beispielsweise habe ich keinen Code für die Auswahl eines ad hoc-Filesystems (z. B. eine RAM-Disk) finden können. So etwas ist für größere Cluster aber unabdingbar, weil Bioinformatikanwendungen sonst im Extremfall ein Filesystem ausbremsen können und damit auch hunderte andere NutzerInnen.
Das ist nach einem Jahr der Einzige bin, der Fehler gemeldet hat oder Fragen hatte, gibt mir zu denken.
Was sagt das über den Review-Prozess?
Mein Traum wäre, das Reviewer sich in dem Gebiet auskennen, dass sie reviewen sollen (z. B. die Welt der Workflow-Systeme) und andernfalls den Review ablehnen. Noch besser die Editoren wählen die Reviewer so aus, dass diese ihrer Aufgabe nachgehen können. Bei einer Software könnte man auch den Test einschließen: Läuft die Software auch?
![]()

Kommentare (3)