
Die Suche nach neuen Wirkstoffen gegen Krankheiten ist eine langwierige, mühsame und teure Angelegenheit. Innovationen gibt es, aber längst nicht in so schneller Folge wie noch vor einigen Jahrzehnten – jedenfalls wenn die moderne Biotechnologie außer Acht gelassen wird und man den Blick auf Wirkstoffe im Sinne einzelner chemischer Moleküle lenkt.
Zur Findung neuer Wirkstoffmoleküle verwendet man u. a. die Methode des Hochdurchsatz-Screenings, bei dem wirklich viele Substanzen, durchaus Millionen verschiedener Moleküle, auf Bindung an Protein-Ziele oder Veränderungen von Zellen getestet werden. In der universitären pharmakologischen Forschung kommen diese recht teuren Verfahren eher selten oder in etwas kleineren Maßstab als in der industriellen Forschung zur Anwendung. In jedem Fall jedoch kann auch auf Computerverfahren zurückgegriffen werden, mit derer Hilfe Substanzklassen eingegrenzt werden können.
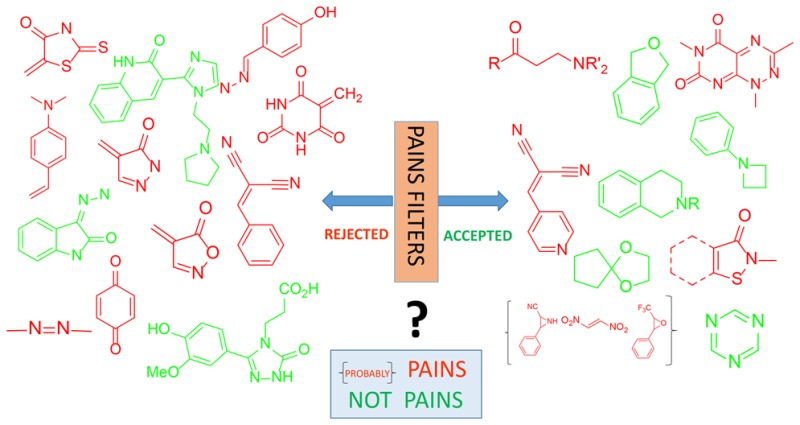
Wie bei allen biochemischen oder informatischen Methoden gibt es einige Hürden, die bei jedem Projekt überwunden werden wollen. Oder auch wiederkehrende Ärgernisse, die es zu bezwingen gilt. So hat vor gut zehn Jahren Jonathan Baell die sogenannten PAINS postuliert (Pan-Assay Interference Compounds)[Baell & Holloway, 2010]. Dies sind Substanzen, die sehr häufig als vielversprechende Substanzen in den Screening-Verfahren auftauchen – und zwar egal, welches Ziel erreicht werden soll. Moleküle also, die zu häufig irgendwie gut binden (oder reagieren), aber unspezifisch. Damit sind sie unerwünscht, weil ein nicht zielgerichtet bindender Stoff keinen therapeutischen Effekt oder aber kolossale Nebenwirkungen haben wird.
Nur wie zum Teufel kann man diese Stoffe ausfiltern? Damit sie auf keinen Fall Tests ruinieren oder gar langwierige Folgetests nach sich ziehen. Und selbstverständlich alles unter der Annahme, dass diese Stoffe keine vielversprechenden Kandidaten für ein Medikament sind. Einfach eine Liste führen? Oder mittels Computer vorselektieren, so dass solche Substanzen erst gar nicht weiter beachtet werden? Ja, wenn das so einfach wäre …. Baell selbst schreibt (eigene Übersetzung nach Baell und Nissink, 2018):
Die Erstellung der PAINS-Datenbank basiert allen auf der (eigenen) Beobachtung – und wurde nicht aktiv hergeleitet von bekannten Toxicophoren (Anm.: Molekülteile, die Moleküle zu Giften werden lassen) oder anderen chemischen Gruppen, die unerwünschte physikochemische oder pharmakokinetische Eigenschaften aufweisen. … es wäre also falsch anzunehmen, dass ein PAIN schlechte pharmakokinetische Eingeschaften aufweist oder jedes Toxicophore ein PAIN wäre.
Das ist genau der Punkt! Kleine Veränderungen an Molekülen können völlig andere Eigenschaften ergeben. Ein PAIN-Teil in einem Molekül, muss das Molekül nicht zwangsläufig zum PAIN machen. Und da auch nur eine recht kleine Menge an Substanzen untersucht wurde, als die PAINS erstmals postuliert wurden nimmt es doch Wunder, dass sie so häufig in der Literatur auftauchen. Tröstlicherweise wird das Konzept auch kritisch diskutiert. Und dennoch möchte bei eigenen Experimenten niemand zu viele falsch positive Kandidatensubstanzen erhalten – und umgekehrt nicht versehentlich geeignete Kandidaten herausfiltern.
An mich wurde die Frage herangetragen, ob man einen PAINS-Filter nicht in einem Computer-Screeningworkflow einbeauen könne (siehe einen früherer Artikel im Blog) und ich war völlig ahnungslos. Als Nicht-Pharmazeut hatte ich davon nie gehört, ein paar Lesestunden weiter schälte sich ein Bild heraus:
Selbstverständlich wird nicht auf Basis ganzer Moleküle gefiltert, sondern auf Basis von SMARTS (SMILES arbitrary target specification) mit deren Hilfe man Molekülteile als eine Zeichenkette definieren kann. Schnell habe ich zwei Listen finden können, welche PAINS als SMARTS definieren. Es gibt noch mehr, doch hier sieht man schon
- völlig unterschiedliche
- nicht gewartete
- nicht annotierte
Inhalte. Ach ja, und Webapplikationen (lies: Blackbox, bei der WissenschaftlerInnen nicht anwenden sollten, weil überhaupt nicht klar ist warum etwas herauskommt und was) gibt es natürlich auch (Beispiel).
Und da fängt der Ärger an. Nicht IT-affinen Wissenschaftlern ist es manchmal schwer zu vermitteln – glaubt mir oder nicht – das eine Software nicht mehr verfügbar ist, weil auf der Seite steht ja, dass sie das ist. Oder gar, dass sie nicht verlässlich arbeiten wird (obwohl es dazu doch ein Paper gibt!1!!). Nachdem man das geschafft hat (also, das Erklären) kommt man bei der Suche auf nach Softwarelösungen auf ein Script wie das oben verlinkte, dass eine nicht näher erläuterte Liste von PAINS zum Input nimmt …
Unabhängig von meinem immer währenden Softwarelamento gibt es grundlegendere Probleme: Dadurch, dass hier irgendwelche Molekülteile irgendeines Tests als Grundlage dienen, kann in einem Screening mit hunderten Millionen von potentiellen Wirkstoffen im Computer natürlich eine große Zahl von Molekülen fälschlicherweise vorab ausgefiltert werden. Einige Brainstorming Sessions und viel Überzeugungsarbeit weiter steht ein Kompromiss: Am Ende des Workflows wird eine Spalte eingeführt, die alle PAINSartigen Kandidatenmoleküle mit einem entsprechenden Warnhinweis versieht. Denn, wie oben schon geschrieben: Es kommt auf den Kontext des Experimentes an, auf das therapeutische Ziel, ob ein PAIN ein PAIN ist – oder vielleicht ein vielversprechender Kandidat, der durch eine winzige Modifikation zu einem spezifischen Binder wird. Oder das PAIN-Molekül weist auf ein “Schwestermolekül” in der Datenbank, das man vielleicht zuvor übersehen hat.
In allen Fällen bleibt: Vorsicht beim Design des Experimentes und des Inputs wird sich auszahlen. Blindes Vertrauen, auf das was irgendwer ins Internet stellt nicht ist wissenschaftlich leichtsinnig. Ich frage mich, wie viele ähnliche Kollaborationen weniger vorsichtig vorgegangen sind …
![]()

Kommentare (3)