
Zur Zeit versuche ich zu erheben, welche Qualität in der Bioinformatik publizierte Software tatsächlich hat. Rein subjektiv hat sich in den letzten 10 Jahren wenig verändert. Das ich meine Zweifel habe, ob die Qualität gut ist, dürfte Euch schon aufgefallen sein (ich denke nicht, dass alles schlecht ist!). Andere haben prinzipielle Bedenken, in Hinblick auf die Qualität wissenschaftlicher Software. Doch behaupten kann man viel. Es gilt erst einmal Daten zu sammeln.
Zum Glück haben das Andere zu einem Teilaspekt schon getan. In diesem, etwas längeren, Artikel möchte ich das aufgreifen. Achtung: Es können hier nicht alle wichtigen Aspekte berücksichtigt werden. Bitte berücksichtigt das in der Diskussion.
Noch eine Kleinigkeit vorweg
Ab und an halte ich Kurse zur Nutzung unserer Cluster. Manche der Teilnehmenden (Studierende ab Bachelor- oder Masterniveau, DoktorandInnen, PostDocs, GastwissenschaftlerInnen, ProfessorInnen, etc. – aus allen Fachbereichen), wissen bis zu dem Zeitpunkt nicht, was ein “Compiler” eigentlich ist. Da stelle mer uns jetzt janz dumm, und sagen so: Ein Compiler, das ist ein Programm, dass Code in (hoffentlich) optimierte Befehle für einen Computer verständlich übersetzt, so dass ein ausführbares Programm herauskommt. Interpreter sind eigentlich ganz ähnlich wie Compiler: Sie legen den Programmtext nicht übersetzt ab (als “Programm”), sondern interpretieren bei jeder Ausführung aufs Neue. — Das soll erst einmal reichen. Wenn Interesse besteht kann ich mal mehr über die Funktionsweise von Compilern oder Interpretern schreiben (aber irgendwie denke ich das ist Teil des Informatikstudiums und wird wenige interessieren).
Doch was ist der Unterschied?
Erst einmal: Kompilierte* Programme können prinzipiell schnellere Ausführung garantieren als interpretierte Programme (oft auch Skripte genannt), weil die Compiler ja a) bereits für den Computer übersetzt haben und b) auch in der Lage sind manche Optimierungen während des Kompilierschrittes einzubringen. Interpreter kennen für diesen Zweck just in time compiler, diese übersetzen und optimieren das Programm (oft auch nur Teile) zur Laufzeit. Theoretisch benötigt ein kompiliertes Programm auch weniger Arbeitsspeicher als eine gleichförmige Lösung, die für einen Interpreter geschrieben wurde: Der Interpreter benötigt an sich schon Speicherplatz und die internen Datenstrukturen sind oft komplexer, während die kompilierten Sprachen sehr genaue Kontrolle über den Speicher erlauben.
Interpretierte Sprachen sind nicht nur in der Bioinformatik, nicht nur irgendwo in der wissenschaftlichen Programmierung, sondern allgemein beliebt. Ein Grund ist der read-eval-print loop (REPL): Man kann interaktiv programmieren und sieht das Ergebnis unmittelbar, ohne jedenfalls den Umweg über einen Compiler. Hinzu kommt im wissenschaftlichen Bereich, dass interpretierte Sprachen i. d. R. auch Bibliotheken an Bord haben, die gute wissenschaftliche Darstellungen (auch “Plots”) ermöglichen. Und Visualisierung ist wichtig!
Wie wirkt sich das im wissenschaftlichen Alltag aus?
Mir geht es um die Qualität wissenschaftlicher Software, insbesondere in der Bioinformatik, da kann ich meinen Vergleich auf die beliebtesten Sprachen in der Bioinformatik beschränken:
Interpretierte Sprachen, also Programmiersprachen, die ein Interpreter zur Ausführung gegeben werden, sind u. a.:
Compilierte Sprachen, also Programmiersprachen, deren Code erst durch einen Compiler übersetzt werden muss, um ein ausführbares Programm zu erhalten, sind u. a.:
- C
- C++
- und seid Neustem gehört auch Rust in die Liste der beliebteren Sprachen in der Bioinformatik, aber da weiß ich nicht, ob der Trend anhalten wird.
Vielleicht kennt Ihr die eine oder andere Sprache und fragt: Und die Shell, z. B. bash? Die ist fraglos wichtig – auf Anwender und Entwicklerseite, aber darin werden keine Anwenderprogramme für WissenschaftlerInnen geschrieben. Und Java? Ist irgendwie ein Zwitter: Mit Aufkommen des “just in time” Compilers in der Java-Virtual Machine (VM) wird nicht mehr “nur” interpretiert, doch andererseits gibt es kein ohne Java-VM ausführbares Programm. Die Veröffentlichung, auf die ich mich beziehe, hatte das Ziel Programmiersprachen in der Bioinformatik zu vergleichen. Sie ist immerhin schon 10 Jahre alt und listet zusätzlich C#, aber die Sprache ist mir egal, weil ich unter Linux arbeite (HPC cluster arbeiten z. Zt. ausschließlich unter Linux und zusätzliche Laufzeitumgebungen, wie mono, machen die Software nicht schneller oder leichter zu nutzen). Und Rust nicht nicht aufgeführt, man kann nicht alles haben. (Wenn jemand eine aktuellere / bessere Veröffentlichung kennt: Her damit!) Hierbei greife ich mal zwei Abbildungen heraus:
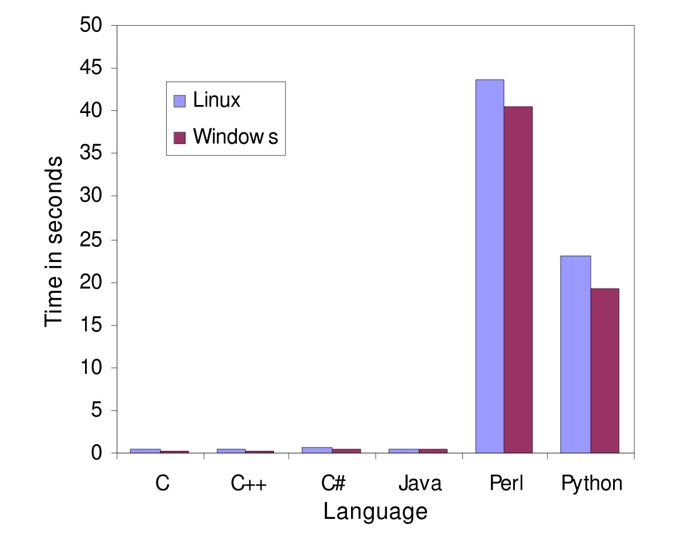
Geschwindigkeitsvergleich bei einem globalem Alignment; es werden also die Geschwindigkeiten bei einer Aufgabe verglichen, die in der Bioinformatik häufig vorkommt. Die Autoren geben an, stets gleichförmig implementiert zu haben. Lizenz: CC BY 4.0
Zwei Dinge werden unmittelbar deutlich:
- die beiden interpretierten Sprachen (Perl und Python) lösen das Problem deutlich langsamer als die anderen Sprachen.
- Linux scheidet konsequent schlechter ab.
Und weiter:
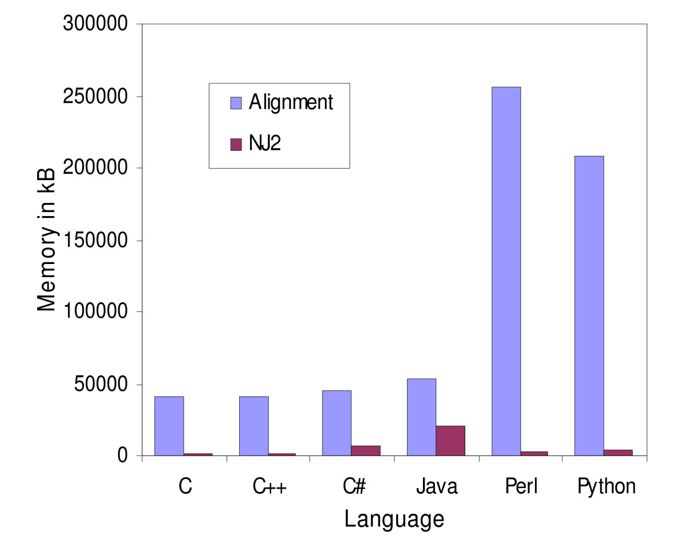
Die zweite Abbildung zeigt die Spitzen des Speicherverbrauchs beim gleichen Problem “Alignment” und bei einem bestimmten Gruppierungsverfahren. Lizenz: CC BY 4.0
Auch hier sieht man: Die interpretierten Sprachen benötigen deutlich mehr Speicher.
Über Benchmarks
Ein Disclaimer: Das Bonmot “Traue keiner Statistik, die Du nicht selber …” kennen wir alle. Bei Benchmarks, also dem systematischen Vergleich zwischen Programmen, Algorithmen, Implementierungen (also Umsetzungen in “Code”), usw. oder eben Programmiersprachen, verhält es sich ähnlich: Sie wecken schnell inneren Widerstand, weil schnell Gedanken aufkommen wie “die Leute haben X nicht berücksichtigt oder Y falsch gemacht”. Das liegt daran, dass eigene Erfahrungen und Wunschvorstellungen den Befunden häufig widersprechen. Womöglich ist der ein bestimmter Benchmark wirklich nicht gut. Aber man kann ja dennoch etwas daraus lernen.
Also weiter …
Der Vergleich “schummelt” ein wenig. Das Problem ist natürlich die Umsetzung: Wenn ich eine Aufgabe in Sprache X gut und in Y naiv umsetze, schneidet Y im Vergleich schlechter ab. Gute Programmiersprachenvergleiche setzen daher auf Streuung: Mehr ProgrammiererInnen, die sich jeweils eine Sprache, in der sie sich gut fühlen auswählen und damit Lösungen erstellen. Und solche Vergleiche sind deutlich differenzierter. Die Autoren obiger Veröffentlichung schreiben “All the programs examined here were written by the same programmer with different levels of experience in Java, Perl and C++. The other languages were implemented while learning them. …”. Da kann und sollte man Fragezeichen hinter manche Details setzen. Aber der allgemeine Trend — kompilierte Sprachen schnell, interpretierte langsamer und speicherhungrig — ist dadurch (für mich) nicht in Zweifel gezogen und ich finde das gerade auch nicht so wichtig: Mir geht es darum Unterschiede bei alltagsnahen Problemen aufzuzeigen – also alltagstauglich für programmierende BioinformatikerInnen. Und zu diskutieren.
Meine Erfahrung – leider (noch) nicht systematisch erhoben – ist:
- Java schneidet in puncto Speichernutzung oft richtig, richtig mies ab. Es gab in der Vergangenheit bei uns sogar Leute, die mehr als 80 GiB pro Rechnerkern (Core) veranschlagten – und das bei vielen, vielen Rechnungen**. Allerdings gibt es auch bessere Java-Programme in der Liga, die mit kompilierten Programmen mithält.
- PyPy, der just-in-time Compiler für Python, ist auch keine Lösung. Mag sein, dass Python so sehr viel schneller gemacht wird. Aber das Risiko die Geschwindigkeit mit erheblich höheren Speicherverbrauch zu erkaufen oder / und mit nicht portierbaren Lösungen dazustehen ist groß.
- Cython, ein Weg Pythoncode tatsächlich zu kompilieren und zu beschleunigen wird selten genutzt.
- Python wird oft derart mies umgesetzt, dass ich mich schon gedrängt gefühlt habe Hinweise zu geben, wie es besser geht – ohne, dass wir auf unserem Hochleistungsrechnern überhaupt Python als sog. Produktionscode (also Skripte, die den Kern von Berechnungen bilden und häufig laufen) sehen wollen – sie sind nicht nur Verschwendung teurer Resourcen, es ist auch unfair, wenn das langsamste Skript anderen Nutzern Rechenzeit nimmt. Um die Bioinformatik mal zu entlasten: Bei uns sind es auch theoretische PhysikerInnen, die schöne Algebra nutzen, diese 1:1 in Python gießen — und sich dann wundern warum die Skripte sooo langsam sind.
- Linux ist selbstverständlich nicht langsamer – das scheint mir eine Auswirkung der Compilerwahl und Einstellungen zu sein. Das Paper bleibt vage.
- Perl kommt in neueren Projekten nur noch selten vor. Es scheint an Bedeutung verloren zu haben.
- R fehlt in dem Vergleich völlig. Diese Sprache wird bei uns einfach so benutzt oder R-Pakete, wie Bioconductor, also eine Bibliothek, die viele Lösungen schon enthält. Bei R und insbesondere Bioconductor ist viel in C oder C++ implementiert und R dient nur als Schicht, die die AnwendungsprogrammiererInnen nutzen – mal kann man also sehr gute Performance erreichen, mal kann man Pech haben und eine naiv implementierte Bibliothek verwenden, die einen Flaschenhals mitbringt. Auch R bietet einen just-in-time Compiler, der bei uns allerdings selten genutzt wird.
Wieso ist das wichtig?
Meine Auswahl der Sprachen und Kenntnisse ist nicht willkürlich. Andere denken auch, dass diese Sprachen und zusätzlich Shellkenntnisse wichtig sind. Natürlich kann man über Details diskutieren. Und solchen Vergleichen wohnt immer der Funke zum Flamewar inne. Also, erst mal durchatmen …
Es ist schließlich egal, ob ein Problem in 2 Sekunden oder 2 Stunden gelöst wird. Ja, das ist für die meisten Probleme wirklich völlig gleichgültig. Und C oder C++ stellen die meisten BioinformatikerInnen vor große Hürden. Warum das so ist, weiß ich nicht. Aber da ist es naheliegend, zu Skriptsprachen wie Python oder R zu greifen. Zu einem echten Problem wird das erst bei richtig großen Datenmengen, wenn also eine Rechnung nicht ein paar Mal, sondern Millionen Male durchgeführt werden muss:

Meine “Motivationsfolie” zum Hochleistungsrechnen. Bild: gemeinfrei, Idee: Lennart Martens
Zum Problem werden interpretierte Sprachen auch, wenn Sie Lösungen verhindern, z. B. weil bereits eine “Lösung” vorliegt, die zwar langsam ist und nur proof-of-concept, also nicht mit allen Eigenschaften (features) versehen, die man braucht. Eine neue, bessere Implementierung wird selten finanziert. Und so vertrete ich die Auffassung, dass vor Start eines Programmierprojektes u. a. überlegt werden sollte:
- Soll mein Programm einmal von Dritten genutzt werden? Nein? Dann sind viele weitere Entscheidungen “Privatsache”. Ja? Dann gilt es sorgfältiger zu überlegen. Nicht vergessen: Der Fokus hier ist wissenschaftliche Software, die veröffentlicht werden soll. Eigentlich selbstverständlich wird aber oft vergessen.
- Wer entwickelt eigentlich? Ich im stillen Kämmerlein? Meine Kollegen mit mir? Eine große Gruppe (mich eingeschlossen) über Gebäude, Länder, vielleicht Kontinente verteilt? Das hat zumindest Einfluss auf die Erwartungshaltung, aber auch auf die Codestruktur.
- Wer soll meine Software wie nutzen? Eine Webanwendung unterliegt anderen Kriterien als eine graphische Benutzeroberfläche, als Programme, die auf der Kommandozeile zu nutzen sind.
- Auf welchen Datenmengen soll meine Software arbeiten? Eine kleine Tabelle oder möglicherweise Terabyte von Rohdaten?
- Und weiter: Welche Algorithmen liegen vielleicht schon optimiert vor? Wie kann ich die mit welcher Sprache nutzen? Welche Parallelisierungsstrategien kann ich damit verfolgen?
Die Liste ließe sich lange fortsetzen. Aber vielleicht dämmert es Euch schon, warum meine Kollegen und ich die Auffassung vertreten:
- Interpretierte Sprachen sind toll, man kann damit viel machen. Insbesondere kann man langsamen und schnellen Code schreiben. Schnellen Code zu schreiben ist ähnlich aufwendig, wie gleich schnellen, zu kompilierenden Code zu schreiben (oft auch unmöglich). Insbesondere leiden Parallelisierungsstrategien in interpretierten Sprachen an ihren engen Grenzen.***
- Daher sollten interpretierte Sprachen für Aufgaben genutzt werden wie Schnittstellen (Wrapper) zu anderen Programmen bereitstellen, Daten graphisch aufzubereiten (zum Plotten), einen Workflow zu skripten, etc..
Persönlich bin ich kein Anhänger des “interpretierte Sprachen haben im Hochleistungsrechnen nichts zu suchen”, aber ihr habt hoffentlich heraus gelesen, dass ich für Sorgfalt in der Wahl der Mittel plädiere.
Meine Schlußfolgerung
Tatsache ist, dass sehr viel Bioinformatiksoftware auf der Grundlage von interpretierten Sprachen publiziert wird (Zwischenstand meiner Erhebung: 80-90 %). Und ist eine Lösung einmal da, wird sie auch genutzt. Auch wenn sie langsam, schwer zu installieren ist und noch andere Probleme mit sich bringt. Man kommt halt schwer zu besseren Alternativen. Und das ist ein Problem, weil langsame Lösungen, die millionenfach Verwendung finden auch viel Strom und Zeit der Anwender und Geld für zusätzliche Hardware verbrauchen – unnötig.
Ganz übel ist die Situation dennoch nicht, denn Qualität setzt sich durch. Die in der bioinformatischen Datenanalyse meist verwendeten Programme (in Bezug auf Rechenzeit) sind in C oder C++ geschrieben oder auch R (mit C oder C++ im Hintergrund). Außerdem spielt Python auch eine große Rolle im maschinellen Lernen – auch hier wieder als “Wrapper” um kompilierten Code, teils sogar für Graphikkarten.
Lösung für Interpreter gibt es häufiger – aber die damit “verschwendete Rechenzeit” hält sich in Grenzen, da diese Lösungen dennoch kurze Laufzeiten aufweisen (z. B. für einen kleinen Plot). Und wichtiger als Grundsatzdebatten ist die Möglichkeit wissenschaftliche Erkenntnisse zu gewinnen. Da spielt REPL eine Trumpfkarte: Die Entwicklung von komplexer Software in kompilierten Sprachen kostet weitaus mehr Zeit als vergleichbare Lösungen in interpretierten Sprachen. Die Überlegung “zu welchem Werkzeug greife ich?” ist eine große Herausforderung. Zumindest – und DAS zeigen solche Vergleiche, sollte man sich nicht nur von Moden leiten lassen.
Und zu guter Letzt gibt es inzwischen auch gute Bibliotheken für die Bioinformatik, die das Entwickeln in C++ oder Rust vereinfachen. Und da purzeln auch Anwendungen heraus: Ein Silberstreif am Horizont bioinformatischer Anwendungsentwicklung …
* Ich “kompiliere” mit dem Compiler – ja, das ist inkonsequent.
** Wir haben derartige Rechnungen/Jobs stark eingeschränkt. Auf einem Tier3-System. Nur falls jemand mit HPC-Erfahrung verwundert einwenden will, dass so Etwas bei ihr / ihm ja gar nicht gehe.
*** Ja, es gibt mpi4Py oder Rmpi (bewußt nicht verlinkt), aber das unterstreicht eher meinen Punkt: Langsame Algorithmen / Implementierungen können viel besser skalieren (wenn ich meine CPUs lange nutze, kann das Zufügen von CPUs zu einer besseren scheinbaren Nutzung führen. Aber dennoch ist das a) eine Verschwendung von Ressourcen und b) gibt es bei interpretierten Sprachen viel engere Grenzen, der Skalierbarkeit. Es gibt keinen Fall, wo man damit viele tausend CPUs gleichzeitig nutzen kann.
![]()

Kommentare (50)