

Ganz zu Anfang des Blogs hatte ich ja schon einmal angedeutet, dass ich einen Zusammenhang sehe zwischen schlechter Software und mangelnder Reproduzierbarkeit von wissenschaftlichen Aussagen. Das es um die Entwicklung von wissenschaftlicher Software, gerade in der Bioinformatik, nicht gut bestellt ist, habe ich ebenfalls behauptet. Und gleich im dritten Beitrag kam die Ten Years Reproducibility Challenge zur Sprache (welche gerade erst nochmal besprochen wurde).
Wie ist die Wahrnehmung der Community?
Den Klassiker zu den Ursachen der Reproduzierbarkeitskrise kennen wohl alle[Ioannidis, 2005]. Immer noch lesenswert, doch finde ich sollte man sich klar machen, dass diese Veröffentlichung, welche die Reproduzierbarkeitskrise vielen erst bewusst gemacht hat, im wesentlichen statistisch argumentiert. Die meisten, wenn nicht alle, statistischen Angaben in Veröffentlichungen der Bioinformatik oder mit Hilfe von Bioinformatik beruhen jedoch auf Analysen mit Software.
Es ist also nicht vermessen zu behaupten, dass die Qualität wissenschaftlicher Software eine Grundvoraussetzung für die Qualität vieler Veröffentlichungen in Biologie und Medizin sind. Da bioinformatische Datenanalysen in der Regel mehrere Schritte erfordern ist es notwendig die Abfolge und Bedingungen gut zu beschreiben – sonst ist eine Veröffentlichung nicht nachvollziehbar und mithin nicht reproduzierbar.
Es gibt da einen interessanten Artikel, der sich an dem Versuch wagt den Aspekt der Reproduzierbarkeit von Software in der Systembiologie zu erfassen[Waldemath & Wolkenhauer, 2016]. Gleich in der Zusammenfassung stellen das Autorenpaar fest:
The lack of suitable standards and appropriate support of standards in software tools has led to numerous publications with irreproducible results.
Wasser auf meine Mühlen! Im Weiteren postuliert der Artikel vier der wesentlichen Probleme:
- Fehlen von Standards zur Erhebung von Daten, welches zu eingeschränkter Vergleichbarkeit führe. Aus der Enzymologie fallen wir spontan eine Reihe von Experimenten ein (z. B. Enzymkinetiken), bei denen zwar alle Parameter angegeben wurden, die eine Messung nachvollziehbar machten (natürlich nicht immer). Aber in jeder Veröffentlichung waren das andere. Damit fällt die Vergleichbarkeit eben schwer. In der genomorientierten Bioinformatik ist das kein Deut besser: Oftmals werden Angaben gemacht, nicht immer vollständig.
- Mangelnde Quantität und Qualität der Daten. Oder sogar vollständige Enthaltsamkeit bei der Angabe von Daten. Gab es bereits einen Beitrag zu.
- Mangelnde Offenheit über Daten und verwendete Modelle oder gar
- Fehlende Transparenz bei Methoden, eingesetzter Software, Beschreibung der Workflows oder der Parameterisierung.
Die Autoren geben in der Folge eine ganze Reihe von best practices zur Problembehebung an. Die werden hier sicher einmal Thema. Mir geht es aber zunächst um eine Einschätzung des Problems, insb. Nr. 4. Und da gibt es eine exemplarische Arbeit[Garijo et al., 2013], deren AutorInnen sich mal der Aufgabe angenommen haben: In diesem Artikel wurden eine Arbeit aus der Wirkstoffforschung[Kinnings et al., 2010] (vgl. mein Beitrag zur Wirkstoffsuche bei SARS-CoV-2, welcher methodisch ähnliche Arbeiten beschreibt) bei Tuberkulose dahingehend untersucht, ob Dritte überhaupt in der Lage sind Schritte nachzuvollziehen, denn wie Kinnings et al. schreiben:
“… the methodology may be applied to other pathogens of interest with results improving as more of their structural proteomes are determined through the continued efforts of structural biology/genomics.”
Muss also gut sein. Spoileralert: Na ja, geht so:
Garijo und KollegInnen haben zwei Dinge festgestellt:
- Es kostet ziemlich viel Zeit den Workflow überhaupt nachzuvollziehen:
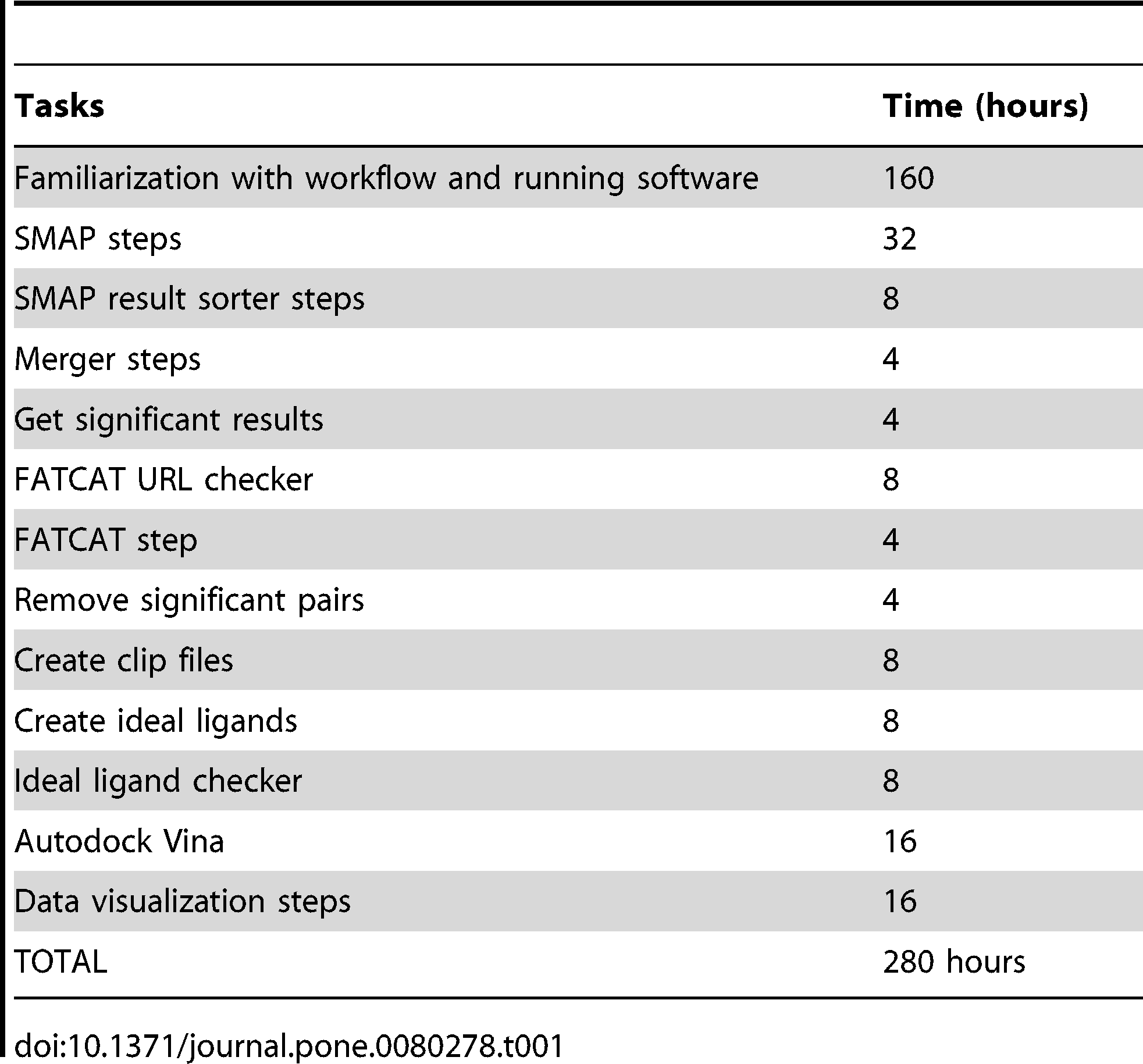
Zeitschätzungen, die es brauchte den Originalworkflow nachzuvollziehen, obwohl Kinnings, also Author der zu grunde liegenden Veröffentlichung, bereitwillig Fragen zu fehlender Konfiguration und Parametern, Programmaufrufen und Validierung der Zwischenschritte beigesteuert hat. (FATCAT ist ein spezifische Schritt, ebenso “Autodock Vina”.)
- Man hat das gleich nochmal systematisch aufgearbeitet. Das war wohl notwendig.
Ok, das ist jetzt ein Teilgebiet der Bioinformatik und dazu noch ein kleiner Ausschnitt eines kleinen Teilgebietes. Das Gros der Bioinformatik ist genomorientiert. Da ist es doch sicher anders?
Ein Schmankerl
Afterwards, the … output was parsed using a custom … script.
Das ist aus einer Veröffentlichung in meinem Umfeld. Wird hier also nicht zitiert, ich will mir keine Feinde machen. Und nein, das custom script, ist auch nirgendwo öffentlich gemacht worden. Solche Veröffentlichungen könnte ich zu Dutzenden anführen. Und in der Regel finde ich plausibel anzunehmen, dass es keine Bewerbungen für Gebiete der Geheimhaltung sind. Das ist also Wissenschaft und die Auslassung ist fahrlässig und ein ernstes Problem. Paper, die versuchen wichtige Workflows zusammenzufassen und allgemeingültig aufzuarbeiten, die gibt es auch in der genomorientierten Bioinformatik. Eine quantitative Erhebung zu dem Thema habe ich allerdings nicht finden können. Vielleicht jemand unter den LeserInnen?
Was tun?
Auch hier gilt: Ohne verbesserte Reviewrichtlinien geht es nicht. Ansonsten, kann man (und ich mache das häufig genug) bei den Autoren nach bohren. Eine Motivation zur Nachbesserung besteht leider selten …
![]()

Kommentare (2)