

Was eine Pipeline ist, weiß jeder …
In der Bioinformatik ist mit einer Pipeline die Folge von Programmen gemeint, die eine bestimmte Analyse mit mehreren Schritten ermöglichen. Hierbei kann die Pipeline, ganz analog einer “echten” auch verzweigt sein. Sprich: Pipelines entsprechen in Ihrer Abfolge von aufgerufenen Programmen stets gerichteten azyklischen Graphen — in der Regel stellen Pipelines in der Bioinformatik jedoch eine lineare Abfolge von Programm-Aufrufen dar, die Dateien lesen und für das Folgeprogramm eine Ausgabe schreiben. Und derartige Pipelines gibt es viele: wer nach ‘bioinformatics pipelines github’ mit einer Suchmaschine fragt, erhält verdammt viele (auch redundante) Ergebnisse und Omicstools (kommerziell, aber man kann ohne Kosten suchen), hat einige hundert registriert.
Ein wenig Hintergrund
Die Idee der Pipe(line) kennen vielleicht manche von Euch von Unix/Linux, wo auf der Shell derartige Kommandos möglich sind:
$ wc -l "$fname" | cut -d ' ' -f1
Dieses Beispiel fragt ab, wie viele Zeilen es für eine Datei die mit der Variable ‘$fname‘ bezeichet wird gibt. Statt einer Ausgabe auf einem Terminal landet das Ergebnis der Pipe (das Symbol für die Pipe in der Shell ist ‘|‘ und das für den Prompt, also die “Eingabeauforderung” das ‘$‘) und wird an das Programm ‘cut‘ gereicht. ‘cut‘ scheidet danach das erste Feld ( ‘-f1) aus und bestimmt seine Felder mit dem Leerzeichen (delimiter ist im Englischen das Trennzeichen, daher der Parameter ‘-d ' '‘. Das ist eine Möglichkeit nur den Zahlenwert zu erhalten, denn sonst enthält die Ausgabe auch noch den Dateinamen.
Der Vorteil einer “Pipe“: Wir schreiben keine neue Datei „auf Platte“, wie hier:
# mit '>' lenken wir eine Ausgabe in eine Datei um $ wc -l "$fname" > temporary_file.dat # mit '<' lesen wir die Datei wieder ein $ cut -f1 -d ' ' < temporary_file.dat
Stattdessen werden die Daten in die „Pipe“ gesteckt, wo sie vom nächsten Programm als Input verwendet werden können. (Unter Unix/Linux ist alles eine Datei, aber das ist mir für diesen Artikel egal. Hier soll eine Datei einfach mal naiv eine gewöhnliche Datei sein, die auf der “Festplatte”, gleich welcher Technologie, geschrieben werden kann.)
Insgesamt ist diese Unix-Pipe(line), die es so natürlich auch auf den vielen Linuxsystemen gibt, sehr mächtig. Sie tummelt sich heute folglich in der Administration von Servern eigentlich überall. Ich versuche das regelmäßig in Bash–Einführungskursen an die Studierenden zu bringen. Und ich versuche auch darzulegen, wo die Grenzen sind, bzw. wo man sinnvollerweise keine Pipeline einsetzt:
$ cmd1 | cmd2 | cmd3
Wenn wir diese drei Programmaufrufe ‘cmd1’, ‘cmd2’ und ‘cmd3’ betrachten, dann gibt es offenbar eine Ausgabe von ‘cmd1’, die ‘cmd2’ zur Weiterverarbeitung gegeben wird und dieses reicht die Stafette weiter an ‘cmd3’. Wenn die gesamte Pipeline insgesamt nur kurz läuft oder alle drei Werkzeuge mit etwa derselben Geschwindigkeit arbeiten, ist das ein sehr mächtiges Werkzeug. Geht es jedoch um viele Daten und beispielsweise ‘cmd2’ viel langsamer als die anderen beiden, so entsteht ein geschwindigkeitsbestimmender Flaschenhals. Im Extremfall ist einige Geduld gefordert. Auf den Teufel, der immer wieder das Detail ausmacht, will ich hier nicht weiter eingehen.
In der “Unix-Welt” haben die gezeigten Pipelines ihre Berechtigung, da die gängigen Kommandozeilentools nach dem Prinzip “Ein Werkzeug, eine Aufgabe” entworfen wurden – bzw. “do one thing and do it well“. So kann man der “Wartbarkeitshölle” entkommen, in die man kommt, wenn man versucht eierlegende Wollmilchsäue zu programmieren. Mal abgesehen davon, dass die Parametervielfalt solcher Wollmilchsäue auch eine erstaunliche Herausforderung für die AnwenderInnen sein kann …
Gibt es da ein Problem?
Wo ist jetzt der Zusammenhang zu Bioinformatik+? Bioinformatik-DatenanalystInnen haben das Konzept „Pipeline“ neu aufgegriffen (wer das Wort neu geprägt hat oder wann, weiß ich nicht → Infos gerne an mich). Hier heißt Pipeline nun: Programm 1 nimmt eine Eingabe, schreibt eine Ausgabe für Programm 2, was diese als Eingabe nimmt und wiederum eine Ausgabe schreibt, vielleicht für Programm 3, usw. — aber die Übergabe geht nicht wie in der Unix-Pipe, sondern als Dateien auf Festplatten (bzw. heutzutage immer häufiger SSDs). Hier wird die Pipeline zu etwas ausschließlich sequenziellen: Ein Schritt wird nach dem anderen ausgeführt.
Die meisten Pipelines gibt es in der genomorientierten Bioinformatik und die hat in den letzten Jahren einen enormen Wandel durchgemacht: Next Generation Sequencing (NGS) ist so “next” nicht mehr und inzwischen sind die Methoden sehr vielfältig. Sogar das Sequenzieren einzelner Zellen, also die Darstellung der Sequenz der DNA einzelner Zellen ist inzwischen möglich. So etwas oder große “Kohortenstudien”, also die genetische Charakterisierung mittels NGS von vielen Menschen zu Vergleichszwecken, generiert viele Daten: Rohdaten von mehreren hundert Terabyte sind keine Ausnahme mehr – und da sind sind nur die Sequenzdaten gemeint nicht die Bilddaten der Sequenzierer.
Wo gestern also noch Programme sinnvoll waren, die genau eine Sache gut machten, sind heute Programme gefordert, die möglichst “File In- oder Output” vermeiden, also nicht immer und immer wieder dieselben Daten lesen und wieder (leicht verändert) ausschreiben: Denn das macht eine Pipeline langsam, auch auf moderner Hardware. Ein wenig Fortschritt hat es gegeben, so waren zum Beispiel vor wenigen Jahren die “FastX”-Programme, die der Entfernung von Sequenzier-Artefakten (für die Fachleute: Das “Adaptertrimming”) und der Qualitätssicherung dienen, ziemlich en vogue – denn das sind Programme, die man gut zu Unix-Pipelines zusammenarbeiten lassen kann. Heute kommen diese Programme in meiner Arbeit als beratender Wissenschaftler nicht mehr vor, außer in der Lehre, wo man damit schön demonstrieren kann, was die einzelnen Schritte machen. Das ist gut so, denn große Datenmengen kann man so wegen des oben angesprochenen Flaschenhalsproblems nicht effizient verarbeiten.
Dennoch hat die Unix-Philosophie des einzelnen Werkzeugs, das seine und nur seine Arbeit gut macht, in der genomorientierten Bioinformatik seine Berechtigung: Da die Komponenten von Pipelines in Arbeitsflüssen (bei verschiedenen Fragen) verschieden kombiniert werden können und sollen, müssen sie eben Komponenten bleiben. Die eierlegenden Wollmilchsäue sind hier somit unerwünscht, weil schwer zu warten und am Ende doch nicht universell einzusetzen.
Und weiter?
Das Zusammenstellen von Pipelines ist eine kleine Kunst. Es gilt:
- Nutzerfreundlich zu sein, also nicht ein Wirrwarr an Optionen zu bieten, die bei der geringsten Fehleinschätzung die wissenschaftlichen Aussagen (die man ja gewinnen will) schmälern.
- Gut zu installieren sollte so eine Pipeline natürlich auch sein – zumindest ihre Komponenten.
- Datei-I/O-Probleme sind zu vermeiden: Ein Tool, das für sich genommen gut und schnell läuft, kann – hundertfach gleichzeitig auf einem System laufend – auch leistungsfähige Dateisysteme ganz schön ins Schwitzen bringen (sofern Filesysteme schwitzen …).
- Schließlich gibt noch die fach- und problemspezifischen Anforderungen.
- Und zu guter Letzt: Portierbar sollte so eine publizierte Pipeline auch sein. Denn sonst ist nicht von Dritten anzuwenden und mithin nicht reproduzierbar.
Zu Punkt 1 kann man als Anwender nicht viel sagen. Hier wird über Download- und Zitierzahlen abgestimmt: Wenn die Pipelines nicht nutzerfreundlich sind, spricht sich das schnell herum. Überhaupt: Die meisten Pipelines werden nicht als solche publiziert, sondern irgendwie für den Hausgebrauch geschrieben. Punkt 2 ist den meisten Anwendern nicht so wichtig, man installiert einmal bzw. schreibt einmal und lässt die Pipeline dann laufen. Und wenn die Pipeline für den Hausgebrauch ist, bestimmt die Installation der Komponenten die “Installierbarkeit” der Pipeline. Punkt 3 ist etwas worüber ich hier sicher mal schreiben möchte, aber im Grunde gibt es auf einem kleinen Server(!) kaum Probleme, die auf Lesen und Schreiben von Dateien zurückzuführen sind, also spielt es für die meisten Entwickler keine Rolle. Punkt 4 ist für unsere Betrachtung irrelevant: Hier geht es mir ums Prinzip.
Bleibt Punkt 5. Doch warum sollte eine Pipeline portierbar sein? Portierbar heißt ja, man kann sie auf einem anderen System einsetzen. Wenn man aber für den Hausgebrauch geschrieben hat, entfällt die Anforderung, oder?
Nun, irgendwann stellen viele BioinformatikerInnen / AnalystInnen fest, dass die Datensätze etwas größer werden. (Womit das Terrain der Softwareplanung erreicht wird, aber wir biegen nicht dahin ab.) Also: Die Pipeline, die so gut auf einem System funktioniert, muss auf ein größeres System. Was tun?

Meine “Motivationsfolie” zum Hochleistungsrechnen — und auch zum Unterstreichen des Rechenbedarfs in der Genomik und anderswo. Bild: gemeinfrei, Idee zur Illustration: Lennart Martens, Lizenz CC BY 4.0.
Option 1: Mehr Rechenpower kaufen. Wenn Option 1 nicht geht (das Budget reicht nicht, die Datensätze sind zu groß, etc.), dann Option 2: In die “Cloud” ausweichen. Ist meist aus Kostengründen und manchmal aus Gründen des Datenschutzes (zumindest wenn es sich um menschl. Genomdaten handelt) nicht möglich (bzw. sollte nicht stattfinden, passiert natürlich dennoch). Und es gibt auch “Bioinformatik-Clouds” auf nationaler Ebene (z. B. de.NBI in Deutschland). Und Option 3 beschäftigt mich: Ausweichen auf den lokalen “Cluster”, als die Systeme zum Hochleistungsrechnen.
Und da schlägt dann ein anderes Problem auf; stellen wir uns einmal vor dies sei eine schematische Pipeline mit ihren Ressourcenanforderungen:
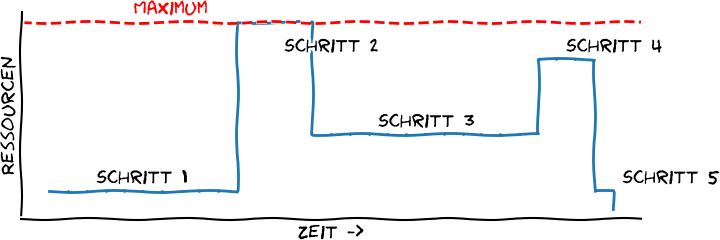
Illustration der schwankenden Ressourcenanforderung auf einem Rechner (z. B. ein 0815 Server oder “fetter” Gamingcomputer) im Zeitverlauf einer Datenanalysepipeline (in der Bioinformatik). Eigene Abbildung, Lizenz CC BY 4.0.
Wenn man das so auf einem Großrechner laufen lässt gibt es zwei grundsätzliche Probleme:
- Läuft die Pipeline genau so als ein Script auf einem Teilrechner (auch Rechenknoten genannt, also ein Rechner mit vielen Kernen und Speicher) des Hochleistungsrechners, gibt es schnell eine Beschwerde des jeweiligen Adminteams: In meinem Beispielplot werden ja im Schnitt nur gut 40% der Resourcen genutzt, wenn das CPUs sind und die Pipeline lange läuft, dann ist das viel Verschwendung.
- Wenn man diese Pipeline so für große Datensätze mehrfach zeitgleich laufen lässt bekommt man definitiv I/O-Probleme (z. B. wegen sog. Random-Access auf Referenzdatenbanken/-genome — das ist auf großen Clustern wesentlich problematischer als auf kleinen Servern. Viele Instanzen eines Programmes können, wenn sie zur selben Zeit laufen, auf ein und demselben Referenzdatensatz zufällig erscheinende Abfragen generieren, weil die eine Instanz hier etwas abfragt, die nächste dort und so weiter. So etwas mögen die meisten Filesysteme nicht. Es gibt Abhilfe, aber all der I/O-Kram soll uns hier nicht beschäftigen.).
Also: Eine solche Pipeline 1:1 auf ein anderes System gar einen Cluster zu übertragen ist manchmal schlicht nicht möglich oder zumindest eine schlechte Idee. Um eine Pipeline zu bauen, der guten Durchsatz garantiert, braucht es Lösungsstrategien, die auf ein Zerlegen der Pipeline hinauslaufen. Schließlich: Wenn der erste Schritt in meinem Schema so wenige Ressourcen benötigt und man gezwungen ist die Pipeline mehrfach zu verwenden: Warum nicht gleich Schritt 1 mehrfach zeitgleich laufen lassen? Oh, warte, da gibt es noch Schritt 2! Auf einem Cluster gibt es die Ressourcen dies mehrfach zeitgleich laufen zu lassen, doch auf dem Entwicklungsserver nicht … Müssen also zwei Versionen dieser Pipeline vorgehalten werden? Wie kann man diese entwickeln?
Ausblick
Weiter geht es mit Ansätzen bzw. Anforderungen, die es gibt, um aus einer Pipeline die notwendige Performance raus zu kitzeln, z. B. wenn man auf ein HPC-Cluster portiert bzw. auf irgendwelche anderen Rechner. Und schließlich mit der Einführung von Workflowsystemen zum Erstellen optimierter, reproduzierbarer und portierbarer Workflows zur bioinformatischen Datenanalyse.
Und hier geht es weiter zum nächsten Artikel (2 von 3) über Pipelines und Workflows.
´´´´´´´´´´´´´´´´´
- DIE Bioinformatik gibt es nicht: Das Fach ist schon etwas älter und so gibt es nicht nur die genomorientierte Bioinformatik, sondern auch die Strukturbioinformatik, die proteombezogene, … schließlich all’ die möglichen Applikation in der Omics-Welt und die Kombinationen und Neuerungen in der Systembiologie. Über ein weiteres Beispiel habe ich letztens mal berichtet. Fazit: Wer allumfassende Kenntnisse für sich reklamiert ist ein Scharlatan.
![]()

Letzte Kommentare