
Pipeline oder nicht Pipeline?
Im ersten Teil ging es darum zu charakterisieren was eine Pipeline in der Bioinformatik ist. Hoffentlich wurde klar, dass die Weise wie manche Datenanlyse Pipeline umgesetzt wird, extrem verschwenderisch sein kann. Wer erst einmal soweit ist zu erkennen, dass eine gegebene Pipeline heterogene Ressourcenanforderungen hat und das dies ernsthafte Performanceprobleme birgt, wenn es gilt größere Datenmengen zu verarbeiten, hat schon viel gelernt. Als Nächstes können wir fragen: Soll es überhaupt eine Pipeline mit diesen Vorzeichen sein? Eine mehr oder weniger lineare Abfolge von Programmen, mit heterogenen Resourcenanforderungen? Oftmals fehlt sogar ein gutes Command-Line Interface (CLI), so dass für neue Datensätze der Code im Hauptskript verändert werden muss, aber selbst wenn eine Pipeline nicht so schlimm ist: Kann man wirklich antizipieren, dass diese Pipeline überall (wo die einzelnen Programme der Pipeline installiert sind) zur Zufriedenheit läuft? Wieviel Enttäuschung hat “works on my system“-Code schon erzeugt?
Meine Hoffnung: Wer hier mit liest und eine — statische — Pipeline schreibt, weiß nun um einige der möglichen Probleme+.
Nun gut, behaupten kann man viel. Zunächst sollt ihr wissen, dass solche grausigen Pipelines tatsächlich publiziert werden. Also liste ich mal was ich finde, wenn ich in pubmed “pipeline” suche. Ein paar zufällige Treffer:
- “The Open Targets Post-GWAS analysis pipeline“; den Code findet man hier; Installation geht über eine virtuelle Machine ansonsten nicht konform mit irgendeinem Standardwerkzeug; immerhin: Es gibt releases und der Code wurde in letzter Zeit gepflegt.
- “Data Analysis Pipeline for RNA-seq Experiments: From Differential Expression to Cryptic Splicing.“; Code findet man in der Veröffentlichung(!); Installationshinweise gibt es über die Hinweise zu ein einzelnen Komponenten; man sollte bitte auch RStudio installieren, also interaktiv arbeiten. Interaktivität bei den letzten Schritten der Analyse (z. B. Auswahl von Graphiken) ist unabdingbar – aber für eine ganze Pipeline ist es ein Performancekiller.
- “SINCERA: A Pipeline for Single-Cell RNA-Seq Profiling Analysis.“; Code findet man hier; letzer Update im Code: 3 Jahre zurück; Installation über R, es gibt keine releases.
- “RACS: rapid analysis of ChIP–Seq data for contig based genomes.”; nur eine Beschreibung, es gibt keinen Code: NutzerInnen müssen Komponenten selber zusammenfügen.
- “Bicycle: a bioinformatics pipeline to analyze bisulfite sequencing data.“; Code gibt es über die Gruppenwebseite; Wartungsstatus und Pflege ist somit nicht nachvollziehbar; im Wesentlichen ein Java-Wrapper; kann man über eine Ubuntu-Live-CD installieren …
- “PRAP: Pan Resistome analysis pipeline”; Code gibt es hier; ein grausamer Stil (system call ohne Warten auf Feedback oder Fehlerbehandlung, selbstgemachtes CLI, etc. etc.); gerade erst veröffentlicht, sind die letzten Änderungen auch schon Monate zurück; es gibt keine releases und keine standardisierte Installtionsroutine. Hatte ich ja schon besprochen.
Dies ist eine willkürliche und kleine Auswahl (und es gibt auch noch die verrückte Idee Pipelines in Programme zu stecken, das klang im Blog hier schon mal an). Sie unterstreicht dennoch einmal mehr eine Problematik in der Bioinformatik: Es fehlt an Kohärenz und Qualität bei der Entwicklung. Viele Arbeitsgruppen machen, was ihnen in den Sinn kommt bzgl. Installation, Softwareumgebung, Skalierbarkeit, Performance, etc.. Und Wartung? Die ist Glücksache. Ach, darüber schrieb ich schon …
Pipelines, im Sinn der Bioinformatik, sind hier eine (weitgehend lineare) Abfolge von Programmen mit (meist) heterogenen Ressourcenansprüchen, die in jedem Schritt die Daten für den Folgeschritt ausgeben (mit Ausnahme des letzten Schrittes). “Interaktive Pipelines” betrachte ich hier nicht weiter, wenngleich Leute auf die Idee kommen so etwas zu schreiben, ja sogar, sie auf einem HPC-System oder in der Cloud laufen lassen zu wollen. Eine wirklich schlechte Idee, weil weder praktikabel noch für große Datenmengen überhaupt geeignet: Wer möchte bei wirklich vielen Läufen einer Software manuell redundante Eingaben tätigen? Na also!
Nun, aus dem ersten Beitrag zu dieser Miniserie erinnert ihr euch an folgendes Bild:
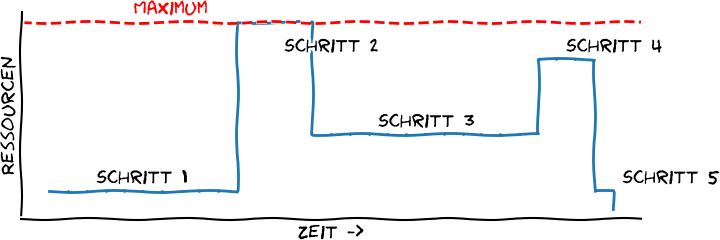
Schematische Ressourcenverwendung einer Datenanalysepipeline. Ressourcen können hier alles sein: CPUs, RAM, …; deutlich wird, dass die verschiedenen Schritte die Ressourcen sehr unterschiedlich nutzen können. Eigene Abbildung, Lizenz CC BY 4.0.
Das ist nicht so gut. Eigentlich müsste es so aussehen, denn nur so stechen die ungenutzten Ressourcen ins Auge:
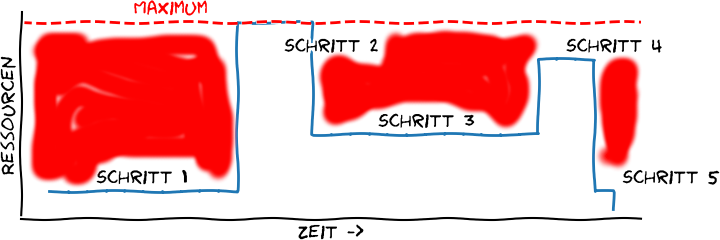
Das “wahre” Bild: ein großer Teil der Ressourcen eines Computers bleibt ungenutzt. Eigene Abbildung, Lizenz CC BY 4.0.
Wie Abhilfe schaffen? – Naiver Ansatz 1
Neue NutzerInnen eines Clusters kommen manchmal auf die Idee: “Ich habe viel mehr Daten, als ich auf meinem Server verarbeiten kann: Lasse ich meine Pipeline (z. B. ein Script) also 1:1 auf einem Cluster (oder auf mehreren Servern mit einem gemeinsamen Dateisystem) laufen. Die Rechenknoten des Clusters sind schließlich “Serveräquivalente”. Visualisiert kann man sich das so vorstellen:
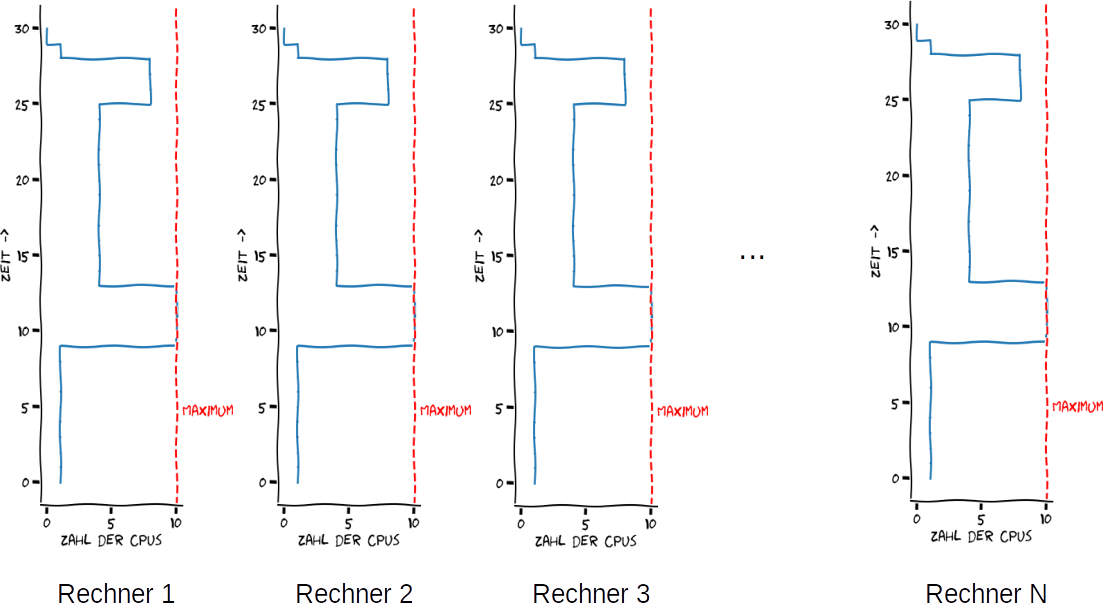
Erster möglicher Ansatz aus mehreren Rechnern die notwendige Performance herauszuholen um unsere hypothetische Pipeline große Datenmengen verarbeiten zu lassen. Hier ist nur die Ressource “Zahl der CPUs” (bzw. Kerne) berücksichtigt und die ist willkürlich auf ein Maximum von 10 gesetzt – moderne Rechner haben selten genau 10 Kerne, meist sind es mehr. RAM und File-I/O bleiben unberücksichtigt. Eigene Abbildung, Lizenz CC BY 4.0.
Warum ist der Ansatz “naiv”?
- viele Ressourcen bleiben im Mittel ungenutzt. Wie viel genau kann man kaum allgemein sagen, aber eine untere Grenze können wir schätzen: Wenn
die Laufzeit einer Pipeline alleine ist,
die apparente Effizienz und
die Zahl der zu analysierenden Datensätze ist die Verschwendung mindestens
.
- Nun deutet die Bezeichnung
- Abgerechnet wird auf einem Cluster nicht die genutzte Ressource, sondern die reservierte. Und weil alle Nutzergruppen um Rechenzeit konkurrieren, vermindert sich der Durchsatz, weil schließlich alle NutzerInnen fair behandelt sein wollen: Wer 40 % der Zeit verschwendet benötigt entsprechend mehr und muss für weitere Rechnungen länger warten. Das ist nicht schlimm bei wenigen Starts der Pipeline, aber bei 10.000 oder mehr …
 die Laufzeit einer Pipeline alleine ist,
die Laufzeit einer Pipeline alleine ist,  die apparente Effizienz und
die apparente Effizienz und  die Zahl der zu analysierenden Datensätze ist die Verschwendung mindestens
die Zahl der zu analysierenden Datensätze ist die Verschwendung mindestens  .
.Und wie sieht so ein Ansatz im Script für ein Cluster aus?
# dies ist an Bash angelehnter Pseudocode, der über # einen bestimmten Input iteriert for sample in ${samples[@]}; do submit-command pipline.sh $sample done
So einfach, das können Anfänger erwiesenermaßen schnell verstehen. Die Batchsystemkommandos können sich selbstredend von System zu System unterscheiden, hier steht nur pseudocode-artiger Shell-Code (und die Existenz von Variablen und Kommandos setzten wir an der Stelle voraus – sie sind nur Platzhalter für das, was Nutzer wirklich wollen).
Ansatz Nr. 2
Wenn nun Studierende bei mir im Kurs aufschlagen, dann haben sie möglicherweise etwas gelernt, sie schreiben ein Masterscript, das in der Lage ist die verschiedenen Analyseschritte in Abhängigkeit voneinander auf den Cluster abzusetzen / zu “submittieren”:
# wieder Pseudocode, der mehr mit "sprechenden Befehlen" # versucht zu arbeiten als mit wirklichen Kommandos. # Der Backslash (\) markiert einen Zeilenumbruch. # Es könnte dort auch eine lange Zeile stehen, die # aber auf der Blogplattform ggf. unleserlich wird. for sample in ${samples[@]}; do jobid_1=$(submit-command step1.sh $sample) jobid_2=$(submit-command --dependency=jobid_1 \ step2.sh $output_from_step1) jobid_2=$(submit-command --dependency=jobid_2 \ step3.sh $output_from_step2) ... done
Und die step-Schritte sind natürlich auch bereits geschrieben und stehen für die Analyseschritte der Pipeline, die die AnwenderInnen durchführen möchten. In der Realität sehen solche Scripte komplizierter aus: Im besten Fall extrahieren sie die Jobid aus dem Rückgabestring des “submit”-Kommandos,
# hier: korrekter Code in bash für das Batchsystem SLURM job_id2=$(sbatch --dependency=afterany:${job_id1##* } \ step2.script $output_from_step1)
sorgen für Übergabe der Dateinamen
# hier: korrekter Code, der die Variablen "input" # und "output_path" als gegeben annimmt job_id2=$(sbatch --dependency=afterany:${job_id1##* } \ step2.script -i ${output_from_step1} \ -o {output_path})
, die korrekte Parameterisierung der einzelnen Schritte
# hier: korrekter Code, der ein paar Variablen \ # zur Parameterisierung aufweist job_id2=$(sbatch -p ${partition} -A ${account} \ --mem=${memory} -c ${number_of_cpus} -t ${time} --dependency=afterany:${job_id1##* } \ step2.script -i ${output_from_step1} -o {output_path})
und im Idealfall auch für Fehlerbehandlung, etc..
Ihr seht: Das kann beliebig komplex werden (und überfordert offenbar das Syntax-Highlighting an dieser Stelle 😉 ). Insbesondere die einzelnen Schritte für die Pipeline können (das ist nicht zwingend so!) Skripte von mehreren hundert Zeilen sein. Schön und wartbar geht anders. Die meisten NutzerInnen überfordert derartiges Skripting völlig.
Doch wie kann man nun die Ressourcennutzung schematisch darstellen?
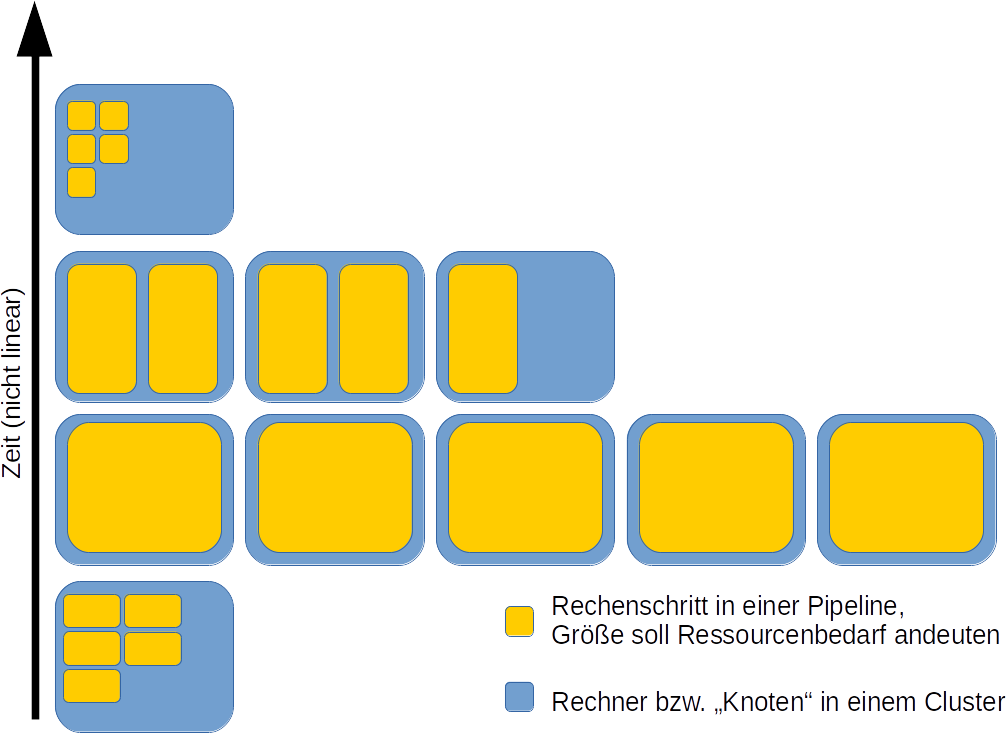
Schematisierte Aufteilung einer Pipeline auf einem Supercomputer (HPC-Cluster) oder in einer Cloudumgebung. Die Zeit ist nicht proportional zu den vorherigen Schemata dargestellt. Eigene Abbildung, Lizenz CC BY 4.0.
Ansatz Nr. 2 nun ist weniger naiv als Ansatz 1 und wird doch selten gewählt. Gut ist der Ansatz ohnehin nur, wenn auch die Skripte step1.sh bis stepx.sh gut sind. Und “gut” bedeutet u. U. für einen Analyseschritt auch ein Skript zur Anpassung einer Software für das jeweilige Cluster zu verfassen.
Aber – hier werden doch auch Ressourcen nicht genutzt, oder? Was ist mit den nicht ausgefüllten blauen Kästen (=Teilrechner) in der Abbildung? In einer Zeit wo der CO2-Fußabdruck von Rechenzentren in der Kritik steht, sind das sehr berechtigte Fragen. Auf einem Cluster allerdings mag es zwar Pausen zwischen diesen Schritten geben (die auch nicht eingezeichnet sind), doch die von unserer Pipeline ungenutzte Resource kann von anderen wissenschaftlichen Rechnungen genutzt werden++.
Wie ist es um die Reproduzierbarkeit bestellt?
In beiden Ansätzen: Nicht gut. Aufmerksamen LeserInnen ist längst aufgefallen, dass wenn eine solche Datenanalysepipeline für ein bestimmtes System geschrieben wurde, es mit der Portierbarkeit auf ein anderes System nicht weit her ist. Und damit ist die Frage nach der Reproduzierbarkeit auch schon geklärt: Sie ist nicht gegeben. Ja, noch nicht einmal die Anwendbarkeit ist sichergestellt. Denn was auf einem anderen als dem Entwicklungssystem nicht genutzt werden kann, ist für Kollegen-WissenschaftlerIn eben nicht zu reproduzieren oder anzuwenden ohne allfällige Änderungen im Code mit allen möglichen Fehlern, die das nach sich ziehen kann.
Wenn nicht eine Pipeline, was dann? Reproduzierbare Workflows zur wissenschaftlichen Datenanalyse in Bioinformatik und anderswo sind gefragt. Wenn der Aufwand zur Anpassung von Einzelprogrammen (die nicht für ein Cluster gedacht waren), hoch ist, bieten Workflowsysteme eine Alternative? Davon mehr im nächsten Artikel, dem Abschluß der Serie.
´´´´´´´´
- Veröffentlicht bekommt man alles. In der (Bio)-Informatik wird nicht selten proof-of-concept-Arbeit veröffentlicht, ohne je die Idee gehabt zu haben diese auch zu warten und Fehler zu beheben. Insofern ist klar, dass das Wissen um Schwächen einer Software (Pipeline oder Programm) viele Leute nicht davon abhält das genau so zu implementieren und zu veröffentlichen.
++ Dies ist ein Grund warum ich es ziemlich kritisch sehe, wenn wissenschaftliche Gruppen größere “Privathardware” anschaffen, statt die föderal bereitgestellten Mittel gemeinschaftlich zu nutzen.
![]()

Letzte Kommentare