
Endlich geht es in der der kleinen Serie (1. Teil, 2. Teil) zu schauen, was zu wirklich nachhaltiger Data Science und damit auch Bioinformatik gehört – und warum das so ist. Zunächst aber: Was haben wir vor Augen haben, wenn wir wissenschaftliche Nachhaltigkeit so richtig weit fassen? Wir können Ziele einer idealen Datenanalyse und alles was mir versuchsweise transparent zu dokumentieren und so zusammenfassen:
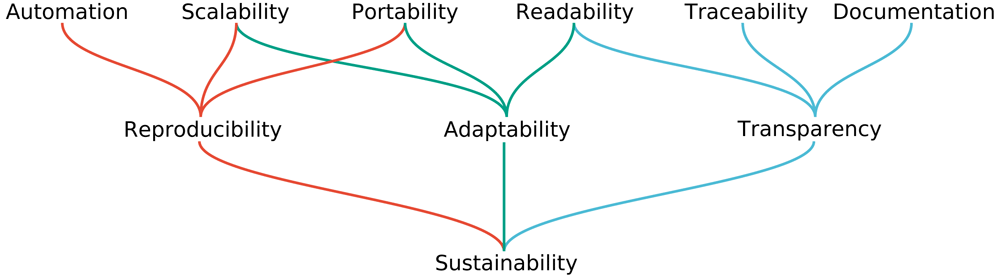
Quelle für diese Abbildung: Mölder F, Jablonski KP, Letcher B et al. Sustainable data analysis with Snakemake. F1000Research 2021, 10:33 (doi: 10.12688/f1000research.29032.1; Lizenz: CC-BY-4.0)
Macht man sich all diese Ziele zu eigen, erreicht man sehr viel: Wir (WissenschaftlerInnen) wollen zunächst einmal Karriere und brauchen dazu Veröffentlichungen. Die meisten – sind wir mal ehrlich – interessiert im Alltag wenig Anderes als eine zügige Veröffentlichung. Dazu wird eine Software benötigt? Na, Hauptsache sie läuft irgendwie. Aber etwas idealistischer sind wir guten WissenschaftlerInnen schon und wünschen uns daher:
- automatisierte Arbeitsabläufe (niemand möchte mit neuem Input jedes Mal manuelle Änderungen im Code anbringen oder viele, viele Male Eingaben per Hand bestimmen),
- einen Arbeitsauflauf bzw. der Code dahinter, der skalierbar ist (es soll egal sein, ob ein oder mehrere Inputs gegeben sind; 100 MB oder mehrere Terrabyte)
- portierbare Arbeitsabläufe (wenn er publiziert ist, sollen Anwender einen Arbeitsablauf auf ihrer Plattform abbilden können, egal ob Workstation oder Supercomputer – natürlich kann man nicht erwarten, dass ein Desktop-PC viele hundert Terrabyte bearbeitet, aber abgesehen davon …),
- lesbares Lösungen, denn wer kann schon seinen eigenen komplexen Code nach 3 Monaten lesen, wenn man nicht sorgfältig ist
- alle Schritte der Datenanalyse sollen nachvollziehbar sein (was passiert wenn sich ein Fehler eingeschlichen hat? Ist dann alles so klar, das klar erkannt wird, wo der Fehler geschah?)
- gut dokumentiert ist (schließlich ist die Computerei egal: Wir wollen ja wissenschaftliche Ergebnisse publizieren und nichts, absolut nichts hält dabei so sehr auf, wie ein Durcheinander im (elektronischen) Laborbuch! – außerdem kann irgendwann jemand mal nachfragen, wenn Zweifel an den Ergebnissen keimen – auch da hilft eine gute Dokumentation)
Ein Schritt zurück
In den letzten beiden Artikeln habe ich Pipelines beschrieben, bzw. das was in der Bioinformatik darunter verstanden wird und wodurch Probleme entstehen können. Implizit ist sind derartige Pipelines Verschwendung von Steuermitteln, weil selbstverständlich unnötiges scaling up geschieht (m.a.W. es werden viel mehr Computer gekauft als notwendig, weil man sich wenige Gedanken machen will und niemand so genau hinschaut, wie viele Mittel es bis zu einer Publikation braucht). Zu zeitgemäßer Datenanalytik gehören aber noch ein paar zusätzliche Kriterien, wenngleich ich der Auffassung bin, dass im einundzwanzigsten Jahrhundert bei der Anschaffung von Servern und sonstiger Hardware ein wenig auf Kosten und Energieverbrauch geachtet werden sollte.
Doch der Unterschied Pipelines vs. Workflows ist im Grunde noch immer nicht definiert, weil ja jede Pipeline einen Workflow (in der bioinformatischen Datenanalyse) darstellt. Doch die gerade skizzierten Ziele lassen sich mit ein paar Eigenbauscripten nicht erreichen. Also unternehme ich einen Definitionsversuch: Jeder Datenanalyseworkflow kann als gerichteter, azyklischer Graph dargestellt werden, bei dem jeder Knoten für einen Arbeitsschritt steht. Die Kanten stellen die Verbindung dar, dies ist die Ausgabe von einem Arbeitsschritt und Eingabe für den nächsten, i.d.R. als ordinäre Datei über das Dateisystem.
An dieser Stelle bekommen wir es mit Workflowsystemen zu tun, Programmen, die uns Workflows erstellen lassen. Sie arbeiten datenzentrisch und konstruieren ihren Arbeitsgraphen durch Kenntnis der Eingaben und gewünschten Ausgaben. Durch das Wissen, welche Ausgabe eines vorangegangen Arbeitsschrittes vorliegt, kann das Workflowsystem den nächsten Arbeitschritt anwerfen und damit eine riesige Durchsatzverbesserung erreichen: Wo beispielsweise viele Schritte eines Präprozessierungsschrittes auf sich warten lassen, einige aber fertig sind, kann – freie Ressourcen vorausgesetzt – der Folgeschritt für die fertigen Ausgaben der Präprozessierung erfolgen.
An dieser Stelle ist ein Beispiel gefordert, damit wir uns das besser vorstellen können. Nehmen wir an, wir wären Linguisten und möchten schauen, ob bei einer gewissen Zahl von Büchern das Zipfsche Gesetz gilt. Wir müssen also die Worthäufigkeiten in diesen Büchern zählen, plotten, eine statistische Analyse machen und am Ende – wir sind ja gute Wissenschaftler(!) – auch Zusammenfassen für eine Archivierung unserer Ergebnisse:
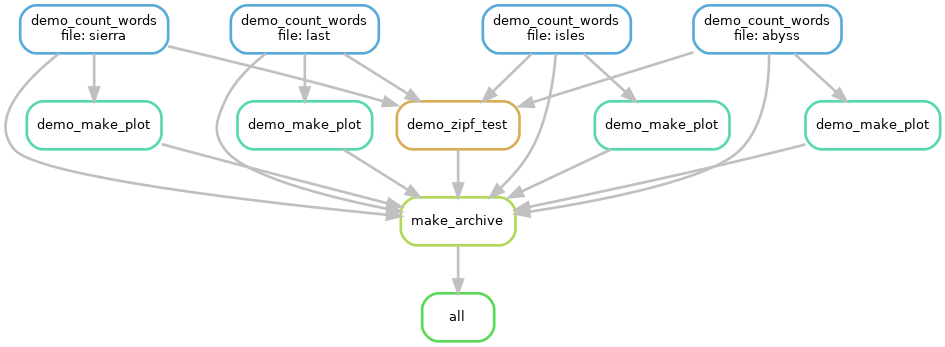
Beispiel, das ich im HPC-Einführungskurs verwende, um in kurzer Zeit (Laufzeit weniger als 2 min) zu demonstrieren, was automatisierte Ausführung bedeutet. Hier werden alle Worthäufigkieten in der Erzählung “My first Summer in the Sierra” von John Muir (hier mit der Datei / file: “sierra”), der Erzählung der Antarktisexpedition von Scott (hier: “last”), von “A Journey to the Western Islands of Scotland” von Keith (hier: “isles”) und in “The people of the Abyss” von Jack London ermittelt. Auf allen Zählungen wird eine Statistik berechnet und auf alle Zählungen werden individuell geplottet (ein Histogramm). Am Ende wird ein Archiv erstellt. Die “all”-Box dient dem Workflow-System zu erkennen was das Ziel ist und ist speziell für das verwendete System. Keine Sorge: Das Zipfsche-Gesetz findet Bestätigung und “the” ist in allen Büchern das häufigste Wort. Was man hier sieht ist ein gerichteter Graph, der im Text angesprochen wurde. Die einzelnen Boxen repräsentieren aufgerufene Skripte und die Bezeichungen darin sind etwas willkürlich, enthalten aber hoffentlich für sich sprechende Namen.
Das Beispiel ist dem Carpentries Incubator entnommen und steht unter CC-BY-4.0 Lizenz.
Klar, so ein einfacher Workflow reicht nur zur Demo. Noch ist der Vorteil nicht recht einsehbar: Was man mit unter zweihundert Zeilen Script erreichen kann, zeigt den Vorteil eines Workflow nicht. Es geht aber auch ein gutes Stück komplexer. So wie eigentlich immer, wenn um wirkliche Analytik geht. Gute Workflowmanager ermöglich auch ein Reporting mit allem Drum und Dran, wenn der Workflow abgeschlossen ist: Zuvorderst natürlich die Auswertung der Ergebnisse, aber auch eine Statistik zum Workflow selber (wie lange hat was gedauert, welche Programme in welcher Version wurden verwendet, etc.) und wer clever ist lässt sich gleich Abbildungen erstellen, die in eine Veröffentlichung kommen. Die folgende Abbildung habe ich der Beispielseite meines favorisierten Workflowmanager entnommen. Für alle, die neugierig auf die Wissenschaft hinter einem Workflow sind, lohnt es sich da herum zu browsen. Hier und heute geht es ja nur ums Prinzip – ja, sogar nur um die Oberfläche des Prinzips.
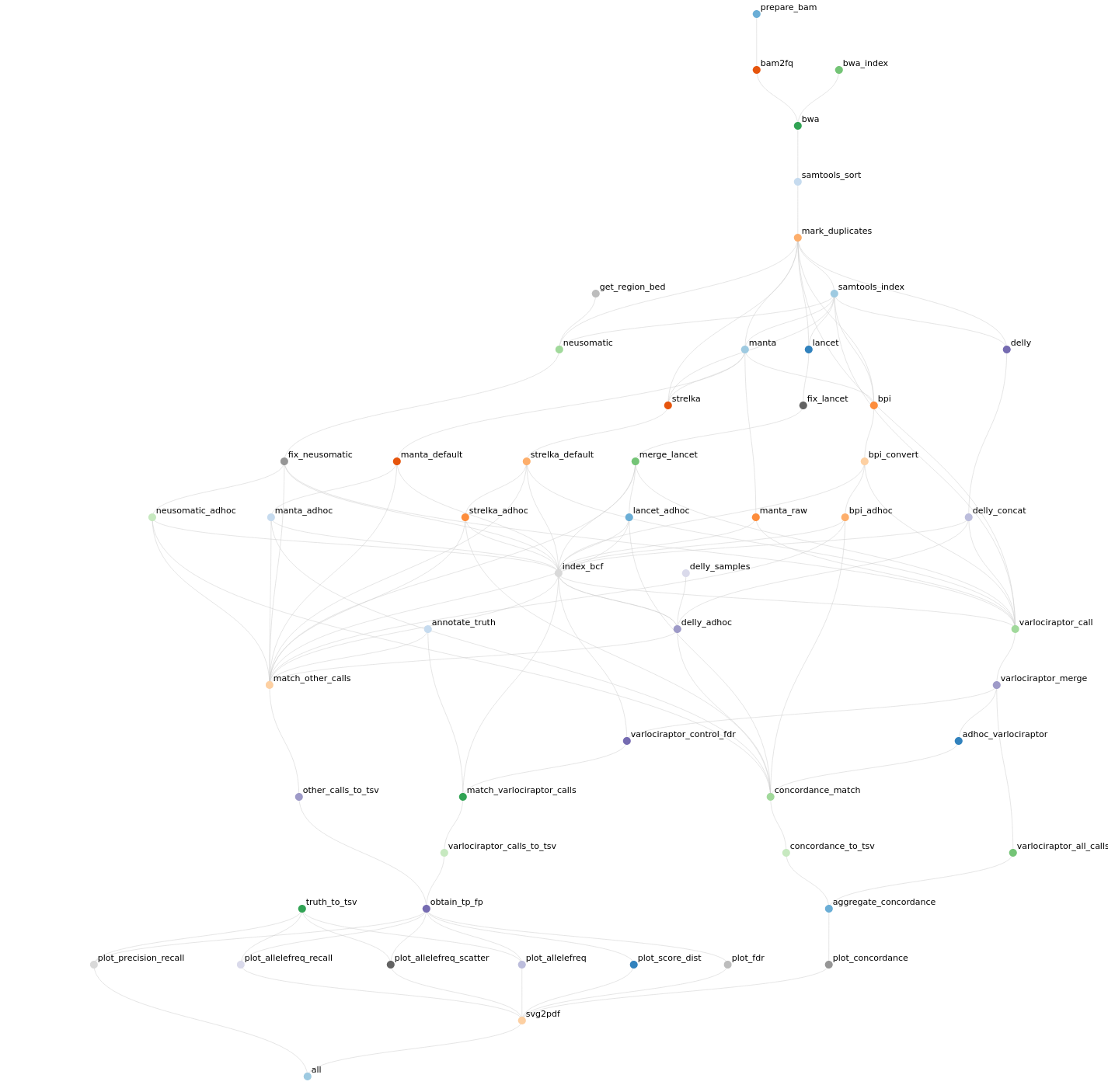
Ein komplexerer Workflow (zur Bestimmung und Klassifizierung von tumorbezogenen Neoepitopen in der personalisierten Medizin). Hier stellen werden die einzelnen Knoten nicht mehrfach aufgezählt (was oben noch pro Input der Fall war), denn dann würde der Graph wahrhaft unübersichtlich. Der Graph zeigt deutlich: Diesen Ablauf per Skript für diese Analyse zu schreiben wäre extrem aufwendig. Das für die nächste Analyse zu wiederholen wäre der erste Schritt ins Burnout.
Derartige Workflowsysteme (bwz. korrekter: wissenschaftliche Workflow Management Systeme) gibt es viele. Nur wenige davon sind weit verbreitet und nicht nicht auf einzelne wissenschaftliche Sektoren eingeschränkt. Es gibt immer wieder mal Versuche zu den etablierten Systemen aufzuschließen – meist ohne Erfolg (Beispiel). Die bekanntesten etablierten Systeme sind Galaxy[Afgan et al., 2008], KNIME[Berthold et al., 2009], Nextflow[Tommaso et al., 2017] und snakemake[Köster et al., 2012; Köster and Rahman, 2012; Mölder et al., 2021]. Die ersten beiden erlauben eine quasi-graphische Programmierung der Abläufe, während Nextflow und snakmake durchaus erfordern selber den Workflow zu scripten (wenn man neu entwickelt) oder zumindest bei neuen Daten eine Konfiguration zu ändern (jedes Mal). Die Wahl kann eine Frage der Vorliebe sein. Mir zum Beispiel gefällt die mangelnde Nutzertrennung in Galaxy nicht und als “Support Scientist” für einen Hochleistungsrechner wäre der Aufwand zu groß. KNIME zu nutzen ist prinzipiell kostenlos, als Service anzubieten kann aber ins Geld gehen.
In einer idealen Welt kann Workflows von einem Management-System in das andere überführen. Bis wir diese erreicht haben, können wir zumindest verschiedene Managementsysteme systematisch vergleichen, bzw. Vergleiche anschauen, die andere für uns gemacht haben. Workflows zu verwenden ist nicht bloß ein Hype, sondern eine begründete Mode: Wo jeder Mensch seine Scripte für ein x-beliebiges System schreibt, liegen Portierbarkeit, Reproduzierbarkeit und Verlässlichkeit in weiter Ferne. Dennoch wird es lange dauern bis selbst gestrickte Pipelines und die Publikation derselben aus der Welt der Datenanalytik und insbesondere der Bioinformatik verschwinden werden und welches der Workflowsysteme sich letztlich wirklich durchsetzt, wird sich zeigen. Letztlich kann es eine längere Phase der Koexistenz geben, doch wo nicht jedes System unterstützt werden kann ist es mit der Portierbarkeit bestimmter Workflows Essig.
Erst gestern wurde mir die Frage gestellt, was es denn braucht, damit Studierende der “Angewandten Bioinformatik” (das mag an anderen Hochschulen unter unterschiedlichen Namen gehandelt werden) am Ende des Studiums denn wirklich bioinformatische Auswertungen machen zu können ohne wie der Ochs vorm Berg zu stehen und bei der beliebten Code-Hilfeplattform stackoverflow Antworten zu kopieren, die sie nicht verstehen. Neben Scriptsprachen (bash, Perl, Python) sind das die Kenntnis eines Workflowsytems und ein Verständnis des Rechners (HPC-System, AWS-Cloud (für die reichen Unis aus Übersee), oder der arbeitsgruppeneigene Server (der womöglich häufig reicht, aber garantiert nicht immer)). In diesem Sinne:
Il computer non è una machina intelligente che auita le persona stupide, anzi, è una macchina stupida che funziona solo nelle mani delle persone intelligente.
Umberto Eco (1932-2016), Schriftsteller, Philosoph und Semiotiker
Der Computer ist keine intelligente Maschine, die dummen Menschen hilft. Im Gegenteil, er ist eine dumme Maschine, die nur in den Händen intelligenter Menschen funktioniert.
![]()

Kommentare (2)